HOPA Publications Committee
Christan M. Thomas, PharmD, BCOP, Editor
Lisa Cordes, PharmD, BCOP, BCACP, Associate Editor
Renee McAlister, PharmD BCOP, Associate Editor
Lydia Benitez, PharmD, BCOP
Alexandra Della Pia, PharmD, MBA, BCOP
Jeff Engle, PharmD, MS
Karen M. Fancher, PharmD, BCOP
Chung-Shien Lee, PharmD, BCOP, BCPS
Robert Steven Mancini, PharmD, BCOP, FHOPA
Bernard L. Marini, PharmD, BCOP
Alan L. Myers, PharmD, PhD
Gregory T. Sneed, PharmD
Diana Tamer, PharmD, BCOP
Kristin Held Wheatley, PharmD, BCOP
HOPA News Staff
David DeRemer, PharmD, BCOP, FCCP, FHOPA, Board Liaison
Michelle Sieg, Communications Director
Joan Dadian, Marketing Manager
View PDF of HOPA News, Vol. 19, no. 3
Board Update: Summer Review and Autumn Preview
Heidi D. Finnes, PharmD, BCOP, FHOPA
HOPA President (2022–2023)
Senior Manager, Pharmacy Cancer Research
Director, Pharmacy Shared Resources, Mayo Clinic Cancer Center
Assistant Professor of Pharmacy, Mayo Clinic Alix School of Medicine
Rochester, MN
Even though summer is winding down, many HOPA initiatives are picking up. Committees turned over in June and their work, along with the work of several Task Forces, has begun. New this year are the Wellness Task Force, focused on combatting burnout, and HOPAmbassadors Task Force, charged with creating an infrastructure for members to become messengers of hematology oncology pharmacy.
HOPA Welcomes Nicole Watts, PharmD, BCOP
Dr. Nicole Watts was recently appointed to the HOPA staff as Director of Strategic Partnerships. Having a hem/onc pharmacist on staff has already begun to yield strong relationships, which we look forward to telling you more about in the coming months. Nicole Watts most recently held a dual position as Clinical Pharmacist in Blood and Marrow Transplantation, and Residency Program Director at the University of Alabama Birmingham.
HOPA in Washington, DC
Practice Management 2022, themed “The Practice of Perseverance,” will occur around the time you receive this issue of HOPA News. That same week, a number of HOPA volunteers will participate in Hill Day, which is returning to in-person! We continue to educate senators and representatives on issues related to oral chemo parity, drug shortages, biosimilars, and the role of the hematology oncology pharmacist and importances of pharmacist recognition as a provider.
Also this month, the HOPA-ASCO 6-Month Quality Training Program will begin with a cohort of HOPA members getting instruction – and gaining hands-on experience – in designing and implementing their own quality projects. Each of the above takes place in Washington, DC, over the course of just five days.
Time to Think Strategically
Recently, the Board of Directors and other HOPA stakeholders took the time to think strategically about the association. Led by 2b Communications & Strategy Group, we brainstormed how to best fulfill our mission of educating hematology/oncology pharmacists and elevating the profession – all while working toward the day that everyone going through cancer treatment has a pharmacist as an integral member of their care team. Enhancements to our Strategic Plan will be released in early 2023.
When it comes to the future of HOPA, as a member, you have a voice! Please vote when the Annual Board Elections take place in November.
Quarterly Town Hall
We have also created a quarterly HOPA Town Hall, where leaders, members, and staff come together to discuss important topics. During each meeting, information will be shared, followed by a live Q&A. Our first HOPA Town Hall was held July 27, 2022 on the topic of Leadership Development and had more 113 registrants! We are appreciative of your submission of topics for our HOPA Town Hall sessions. The next Town Hall is Tuesday, October 18, 2022 and we will discuss HOPA Advocacy efforts.
Did you Know?
- American Pharmacists Month is right around the corner, beginning on October 1! It recognizes the role pharmacists play in our health, communities, and lives. Watch for HOPA’s month-long celebration, including Women in Pharmacy Day on October 12.
- Oral and IV Chemo Education Sheets, in collaboration with NCODA, ONS, and ACCC, are available for download, with more new releases to come!
- HOPA and Pharmacy Times have partnered to create a 12-month video series, featuring HOPA members. Go to pharmacytimes.com and look for us under Resources and then Partners.
Feature: Clinical Trial Management in the Post-Pandemic Era: Reflections of an Investigational Drug Service on the Past Two Years
Jennifer Murphy, PharmD, BCOP
Senior Pharmacist, Oncology & Investigational Drug Service Investigational Drug Service, Cancer Center
Assistant Clinical Professor, UC San Francisco School of Pharmacy
Department of Pharmacy Services, UC Davis Health System
Sacramento, CA
The COVID-19 pandemic has fundamentally changed the operation of clinical trials over the past two years. The extent of the pivot and the rate of change in operations varied amongst clinical trial centers and Investigational Drug Services (IDS) as the COVID waves rippled across the country. While there have been numerous lessons learned by all, this unexpected impetus has altered the landscape of how clinical trials will be conducted in the future as we move into the post-pandemic era.
For those us who practice in an IDS pharmacy or manage patients enrolled in clinical trials, we now begin to reflect upon the past two years. It is important to evaluate which of the new strategies that were implemented hold enough value to continue in the post-pandemic landscape. Many new workflows and approaches were adopted: clinical trial patient telemedicine visits, mailing of investigational product (IP) to the patient’s home, virtual monitor visits, and virtual audits. Practices that were once thought impossible or undesirable were implemented out of necessity. In fact, some of these workflows have become preferred by many due to the conferred efficiencies.
Prior to the pandemic, most IDS pharmacies and trial units hosted their monitoring visits exclusively in person. To reduce exposure to COVID-19 most sites enacted severe visitor restrictions. To balance the regulatory oversight of clinical trials and the risk reduction, clinical trial units and IDS pharmacies sought alternative strategies to fulfill contractual objections. When visitor restrictions were put into place, most sites transitioned to virtual or remote visits. Some sites had already implemented virtual visits prior to 2020, and they served as role models for those of who looked to quickly implement an entirely new workflow overnight. Hosting virtual IDS and regulatory visits provided IDS pharmacies new benefits and challenges. Leveraging technology such as secure meeting platforms, email, telephone, and electronic inventory systems as virtual monitoring locations, staff no longer need spend to their days sitting with their on-site visitors and hosting in-person visits. For those with electronic accountability systems, study documents can be uploaded in secure platforms or provided electronically not only saving time, but immense amounts of paper and physical meeting space. For those managing paper accountability forms and other paper documents in physical binders, it presented the challenge of additional time spent making copies, scanning, and organizing that the monitor would have otherwise done. The benefits of a paperless IDS operations became even more clear given the apparent efficiencies that these remote visits can have. What once seemed to be a temporary process to bridge the gap, now appears to be a strong contender for permanent change in monitoring visit operations at trial sites.
The care and management of clinical trials traditionally has required patients to come on-site for in person visits. During the initial release in March of 2020, the US Food and Drug Administration published guidance for the conduct of clinical trials during the pandemic. Recommendations provided to date state that “alternative methods for safety assessments (e.g., phone contact, virtual visit, alternative location for assessment, including local labs or imaging centers) could be implemented when necessary and feasible.”1 Accordingly, sponsors and Institutional Review Boards (IRBs) issued permissions for telemedicine visits. This allowed patients who were otherwise scared, hesitant, unwell, or required by local ordinances to stay at home to continue to have access to medical care. Virtual strategies employed for COVID risk reduction increased the frequency and use of telemedicine visits for standard of care and clinical trials in early 2020. An article by Marra and colleagues published in June 2021 found that the use of connected digital products only increased by 1.65% between May 2020 through February 2021. While this increased is much smaller than anecdotally expected, the author concludes that the “options created by regulatory guidance to stimulate telehealth monitoring were not widely incorporated into clinical research.”2 Nevertheless, this transition did allow many clinical trial patients to remain enrolled in their clinical trial and continue treatment. Looking at forthcoming protocols, telemedicine visits are included in protocol procedures as alternatives to on-site visits when appropriate. This is a warranted change for many reasons. A model that includes telemedicine visits has the potential to increase accessibility to clinical trials for patients outside the institution’s catchment area, bring the clinical trial to the patient’s home, reduce travel-related costs incurred by the patient, increase diversity of subjects, and increase equity and inclusion of underinsured, underrepresented, and at-risk patient populations. While on-site visits will likely remain the preference to ensure proper oversight and accountability, a hybrid model is likely to remain a viable option when feasible.
To complement the telemedicine visits and assessments, many sponsors provided exceptions to a previous prohibition on shipping IP to the patient at home. Included in this group of sponsors was the National Institute of Health (NIH) and National Cancer Institute (NCI) Cancer Therapy Evaluation Program (CTEP) of the Division of Cancer Treatment and Diagnosis/NCI which temporarily permitted shipment of IP from the site to the patient home during the pandemic. In January of 2022, the NIH/NCI provided notice that “for studies under CTEP IND with oral investigational agents, CTEP will allow the Dispensing Pharmacy to ship oral investigational agents directly to study subjects.” This is a significant change in trial operations and a signal that mailing IP is an acceptable method to manage patients remotely as we move past the pandemic. This change in practice has provided continued access to clinical treatments during the pandemic and will continue to do so as we emerge into a new way of conducting clinical trials.
While the benefits of shipping are clear, there are many challenges associated with shipping IP which may limit the use of telemedicine and mailing of IP in the future. Without patients coming on site for their visits, real-time accountability of patient returned IP is not always feasible. To accomplish this remotely, oral IP accountability will need to occur over a telemedicine visit or through creative methods (e.g. patient submitted photographs, technologically enhanced IP bottle caps that track adherence and doses). Using a phone, camera, or computer may be difficult for patients as they attempt to maneuver their electronic devices close enough for the provider to view an array of capsules or tablets. Formally, IP accountability would need to be done once the IP supplies for the prior cycle(s) are physically at the site. From a site perspective, shipping IP to patients can be time consuming for staff and expensive. Agents with a CTEP IND require the use of a qualified shipper (to maintain protocol required temperatures and conditions) in addition to the cost of shipping the package, which can be costly. There may not always be available funds to cover the shipping costs. Additionally, pharmacy shipment of IND-exempt IP directly to the patient is subject to Board of Pharmacy regulations. Due to the associated licensing and regulatory complications, this often removes the ability to ship this IP interstate and requires the patient to come on site to pick up their outpatient medication(s). Limitations such as these may prevent some sites and patients from taking full advantage of what is otherwise feasible and frequently used dispensing process for standard of care patients. Finally, the additional physical space and labor involved to provide shipping services on a frequent basis cannot be understated. The support of pharmacy departments, health system and research offices must be present to advocate and consider any additional resources that may be needed in the IDS pharmacy.
As we move into the post-pandemic era, the number of clinical trials activated at sites will naturally increase opportunity for enrollment. From February 2020 to May 2020, clinical trial activations “were only 57% of the expected estimate had the pandemic not occurred” as compared with 77% of non-US-based trials.3 While a rebound in activated trials occurred across sites as communities re-opened, many clinical trial sites are still impacted by COVID-related staff shortages and continued restrictions.
If the pandemic serves as nothing else, it has been a catalyst for change. It has provided clinical trial units, IDS pharmacies, clinical trial research organizations (CROs) and sponsors with a new vantage point and a glimpse into what the possibilities are: a research world where the clinical trial meets the patients at their home and increasing efficiencies for those operating the trials through the leverage of technology and modern structure. With eased restrictions and the return to some semblance of pre-pandemic times, healthcare providers, pharmacies, and trial units are faced with the reality of deciding what the future of clinical trials holds. It is up to all stakeholders to evaluate what serves their department, site, patient population, and research community well and to part ways with what does not serve them. Just as the past two years have forced clinical trial units, IDS pharmacies, and supporting departments to operate with the utmost of patient centered focus, the pandemic can also now serve as a reset and a chance to revise and/or reaffirm operations. By embracing these opportunities in the long-term, health care providers, clinical trial units, and IDS pharmacies can improve their efficiencies, improve patient centricity, and practice in line with their values and patient centered models.
REFERENCES
- Food and Drug Administration. (2021). Conduct of Clinical Trials of Medical Products During the COVID-19 Public Health Emergency: Guidance for Industry, Investigators, and Institutional Review Boards. https://www.fda.gov/media/136238/download. Accessed 21 JUN 2022.
- Marra C, Gordon WJ, Stern AD. Use of connected digital products in clinical research following the COVID-19 pandemic: a comprehensive analysis of clinical trials. BMJ Open 2021;11:e047341. doi: 10.1136/ bmjopen-2020-047341.
- Unger JM, Xiao H. The COVID-19 pandemic and new clinical trial activations. Trials. 2021;22(1):260. Published 2021 Apr 8. doi:10.1186/s13063- 021-05219-3.
Jumping into the Pool: A Career Transition into Pharmaceutical Industry
Ashley E. Glode, PharmD, BCOP, FHOPA
Health Systems Oncology Medical Affairs Director
Merck & Co., Inc.
Aurora, CO
Michael Vozniak, PharmD, BCOP, FHOPA
Scientific Director Medical Affairs
Merck & Co., Inc.
Ambler, PA
Attending HOPA’s Annual Conference this past March in person was exciting and energizing! It was great to re-connect with so many colleagues I have not seen or engaged with since Tampa in 2020. Over the two years, one would hear about people making a transition to work in the pharmaceutical industry, but it wasn’t until seeing and networking with people at the Annual Conference that I realized it was a huge trend. Due to the immense volume of cancer research and corresponding development of novel agents and combinations aiming to improve outcomes, oncology pharmacists are exposed to the pharmaceutical industry and the work pharmacists in industry perform. Roles for pharmacists in industry are varied and not contained within one department. Opportunities exist within clinical trial development, drug safety, regulatory affairs, medical affairs, medical writing, corporate education, marketing and sales, to name a few, and may vary based upon the size and scope of the pharmaceutical company. It is not surprising that experienced and highly trained oncology pharmacists are attractive applicants for pharmaceutical industry positions.
In this article, Ashley Glode who made a transition to industry in the past year and I reflect on our careers and our transitions to industry. We hope that our journeys help you, as you consider your next career steps.
What inspired you to specialize in oncology pharmacy?
Glode: I fell in love with oncology as a pharmacy student at Duquesne University. The advancements in pharmaceuticals and need to be a lifelong learner to keep up with the evolving treatment landscape has kept my passion alive. Now I get to work for a company bringing new treatment options to cancer patients and enjoy being on the cutting-edge learning about new potential options.
Vozniak: I was fortunate to have very engaging and dynamic oncology professors at the University of Pittsburgh. While the curriculum was tough, they instilled the excitement and challenges of working in oncology. During PGY1 residency, I was drawn to complex and acutely ill patients and enjoyed not only oncology but also infectious diseases and critical care. As I looked ahead, it was apparent there would always be new advancements in oncology therapeutics to keep abreast of. With this in mind, I decided to pursue PGY2 oncology training and have never looked back. During my PGY2 training, my passion for oncology grew as I observed how the role of the pharmacist was valued by the patient care team and the impact a pharmacist can have on many aspects of patient care.
What was your career path prior to transitioning into pharmaceutical industry?
Glode: Following my PGY2 residency, I knew I wanted to find a position that would combine my interests of oncology, research, critical care, and infectious disease. My first job was as the inpatient hematology/stem cell transplant clinical pharmacy specialist at the Medical University of South Carolina (MUSC). In this role I had a joint faculty appointment at the SC College of Pharmacy/ MUSC College of Pharmacy, was the residency program director for the PGY2 oncology residency, and preceptor to numerous pharmacy residents and students. After 5 years at MUSC working with so many trainees, my passion for teaching led me to a faculty position at the University of Colorado School of Pharmacy with a joint appointment in the Phase I/GI/Head and Neck/Sarcoma multidisciplinary clinic at the University of Colorado Cancer Center. I spent 7 years in this position developing my teaching skills and expanding my knowledge base in clinical trials and solid tumors. In the clinic I was able to collaborate with world renowned providers on improving patient care and various research projects.
Vozniak: Upon completing PGY2 residency, my first job was as an oncology clinical pharmacy specialist at the Hospital of the University of Pennsylvania. During my 9 years in this role, I spent time covering gynecologic oncology, medical oncology, and hematology/oncology including stem cell transplant. To this day, I tell people that this was the coolest and most demanding job that I have ever had. I worked with some of the brightest people in the world and it made me want to do the absolute best I could for our patients and our medical team. I was involved in the customary responsibilities of the role such as formulary review, guideline development and precepting. I then transitioned to be an Associate Director of Pharmacy and helped to oversee our department’s pharmacy practice model. I served as a people manager for the first time and helped to manage over 100 pharmacists. This role helped me see the bigger picture of our department and health-system as our multi-hospital system began to streamline across several areas.
What prompted your career transition into pharmaceutical industry?
Glode: After having a fruitful career at MUSC and the University of Colorado and being able to accomplish many of my career goals, I was itching for a new challenge. I had always thought my career would transition to an industry position at some point, but I wasn’t sure how and when. Feelings of burnout and the pandemic led me to write a pro/con list of staying in my current role vs. making a career change. Not having as much connection with my students, colleagues, and patients, I found myself feeling less fulfilled and began seriously evaluating my career and my identity. I felt isolated and frustrated by things out of my control and was no longer the passionate oncology pharmacist I had once been, or the role model I wanted to be for students and residents. In September, my former colleague had reached out and encouraged me to apply for an opening at Merck in my region. I wasn’t sure in the moment and thought about it for several days before submitting my application. When meeting some members of my team and learning more about the position during the interview process, my excitement grew, and I felt like I had found my “pharmily." I loved the unique aspects of this specific role and the ability to blend my passions of lifelong learning, teaching others, and improving patient care and access to treatment. It felt right in my gut, and the pros outweighed the cons on my list, so I made the leap, and I am very happy I did.
Vozniak: During my time as Associate Director of Pharmacy, I served as HOPA President, and I began to have routine engagement with pharmaceutical industry personnel. I recalled a message a mentor once shared- when you volunteer your time and efforts, be sure that you get something out of the experience; you should grow from it as well. So, I began to ask questions about the roles for oncology pharmacists in industry. This coincided with a time when I was looking ahead and considering my next possible steps in academic pharmacy. I realized that I didn’t want to be a Director of Pharmacy and I should consider a different career path. My growing knowledge about roles in industry and networking connected me to available positions in industry and I found a good match to make the jump.
What do your day-to-day activities look like in your new role?
Glode: As a Health Systems Oncology Medical Affairs Director I provide the most up to date information from major congresses and publications to decision makers and payors, so they have what they need to make treatment access/formulary decisions. I attend regional and national conferences to gather insights on practice and opinions from thought leaders in my territory and across the nation.
Vozniak: I collaborate with several departments in our company to keep informed of updates to both our company products and key competitor products to develop and update our strategy for our field-based team. I share key insights from our field team with internal colleagues to keep them informed of thought leader opinions and reactions.
What advice would you give someone looking to make a change into pharmaceutical industry?
Glode: Whether you are a recent graduate, or an experienced pharmacist, keep in mind these few tips: 1) If you’re unsure you’re ready to make the change into pharmaceutical industry, do not hesitate to apply for a position you are interested in and interview to learn more. 2) If you know you are ready to make the change, networking is key to your success. Don’t be afraid to let your close friends and colleagues know you are interested and use those connections. 3) Everyone’s path is unique, and things happen for a reason. If it is meant to be, it will happen. Just hang in there and use your network to support you on this journey.
Vozniak: Networking helps- oncology pharmacy is a small world and so is industry pharmacy. Getting your foot in the door is tough, but once you get in, it is easier to navigate to other roles within the company. Be willing to take on a job that you think you may be overqualified for. Finally, it can take time. Be patient and continue to build your network in the industry space.
We are employees of Merck & Co., Inc., Rahway, NJ, USA, but we are not speaking today on behalf of the company, the opinions or perspective included or expressed throughout this presentation/discussion are our own and do not represent the opinions or perspective of our employer.
Navigating Payer Mandates and Staying a Step Ahead
Laure DuBois, PharmD, BCOP
Pharmacy Manager for Oncology Services
The University of Kansas Health System
Kansas City, Kansas
Nikki Ogle, PharmD, BCOP
Team Lead Reimbursement Oncology Pharmacist
The University of Kansas Health System
Kansas City, Kansas
Introduction
In the oncology landscape, payer issues can be commonplace. Some of the issues that we face as pharmacists include step edits and site of care restrictions. The goals of these restrictions are to help reduce costs and to improve patient outcomes, but they often impede patient care by slowing down the authorization process, causing confusion for financial teams, and change rapidly. To combat these challenges, our institution has implemented several processes.
Background on Step Edits
Step edits, also known as “fail first” edits, require a patient to try an alternative therapy that is usually less costly before using the therapy that was originally prescribed. In 2018, the Centers for Medicare & Medicaid Services (CMS) announced that they would use step edits for Medicare Advantage Plans in patients with cancer. 1 This was then expanded to include Medicare Part B in 2019. Once this started, many commercial payers followed suit.
When biosimilars came to the market, they presented a new angle for payers to include step edits in their contracts. Many payers have preferred biosimilars that a patient must try and fail before receiving another biosimilar or the reference product.
Key Considerations on Step Edits
There are many considerations when navigating step edits, including patient implications and authorization considerations, stocking and storing, and preventing medication errors.
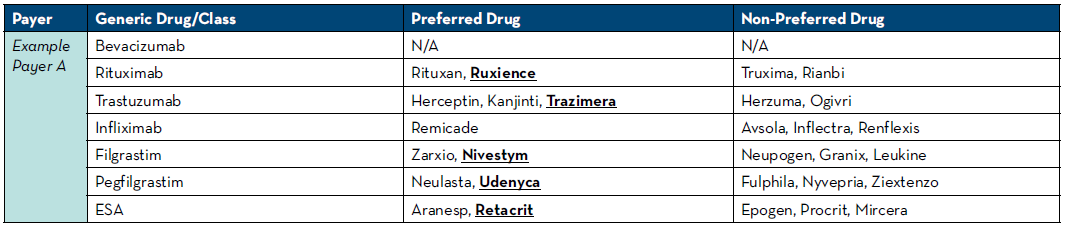
For insurance to cover the cost of an agent, your institution must obtain authorization for use prior to starting therapy. Running into a step edit at the time of authorization may lead to patient delays as this often puts the authorization process back at the beginning when requesting the new preferred agent. We have leveraged technology to assist with this and have rules written in our electronic medication record (EMR) system to look at the patient’s payers and automatically select the known mandated drug. If payers do not have a preference, we default the rule to our institutional-preferred product. To do this, we pull payer guidelines at least quarterly and keep an updated spreadsheet of preferred and non-preferred agents with the payers seen at our health system (Table 1). Another option if rules are unavailable would be to place communication orders into order sets alerting providers of which medication will be approved based on payer. Engaging industry partners is very helpful to know about changes to payer policies as they are usually knowledgeable about their medication being moved to and from preferred and non-preferred statuses.
Another step we implemented was a biosimilar auto-substitution policy. This policy allows our front-line pharmacists to change the biosimilar to the preferred product that we may only learn during the process of obtaining the authorization. We have found that the pharmacist is usually able to adjust these medications and select the right biosimilar in a timelier fashion than the physician in the clinic.
Step edits can also mandate using a generic formulation of a product instead of a brand name product. This can have downstream effects on patients if they were utilizing any form of medication assistance that is not available for generic medications. There are also times when a step edit may not be appropriate for a patient. For example, we have been asked to change pegfilgrastim to a preferred filgrastim product. In these cases, you often must go through the peer-to-peer process to get the agent you are wishing to use approved which can lead to delays in getting patients started on therapy.
Biosimilars are nightmares for look-alike sound-alike errors. To help mitigate these errors, consider only stocking your institutional-preferred biosimilar in your automated dispensing cabinets. Safety tools, like barcode medication administration, are another added layer of defense for these errors. When storing inside the pharmacy, consider placing biosimilars on different shelves and not next to the other biosimilar products. Narrowing down to one preferred agent that is accepted by payers for most patients can be helpful to decrease storing multiple biosimilars.
Background on Site of Care Restrictions
We all dread hearing the words “alternative site of care needed.” There are emerging trends in the oncology world where payers are wanting a lower site of care than a hospital outpatient department (HOD). These site of care restrictions are often targeted at supportive care medications, monoclonal antibodies, complement inhibitors, and immunotherapy.
Table 1: Preferred and Non-Preferred Agent Reference
Key Considerations on Site of Care Restrictions
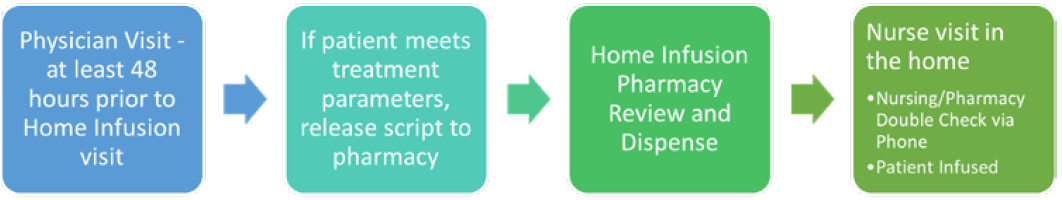
We have implemented utilizing our home infusion pharmacy and/or self-injection when possible. Centers that currently do not offer home infusion services may need to start considering the pros and cons of these services. Many therapies that are not covered in HOD locations are able to be serviced by home infusion. For our cancer care partnership with home infusion, we started small and progressed to more complicated targeted agents. An easy start included medications such as IVIG, antibiotics, and growth factors. As we started to expand to targeted therapies, we made the decision to utilize our EMR system to help us with the process.
We learned quickly that oncologists like to see everything that was occurring with the patient when they transferred to home infusion. We built-out home infusion as their own treatment department with a schedule that is viewable by anyone at the institution to mirror our treatment room in the cancer center. We utilized our order sets that already existed and built-out take-home prescription orders that could be e-scribed to our home infusion pharmacy. This increased satisfaction from the oncology team as they could see where patients were in therapy and were able to visually track their cycles of therapy.
After receiving feedback from physicians and patients, we adjusted some of our home infusion workflows. To allow home infusion time to prepare for a nurse visit in the house with an accurate dose, we implemented at least a 48-hour window from physician visit to administration of the medication in the home. This allowed for dose changes and pharmacy double checks for high-risk medications. To allow for a dual verification of immunotherapy in the home we implemented a process where the home infusion nurse calls the home infusion pharmacy to verify patient, dose, vital signs, and treatment parameters were met prior to infusing the medication (Figure 1).
Home infusion services do have pitfalls. One thing to keep in mind is that most payers will only authorize one site of care, so patients are either in the infusion room or home infusion. Patients with Medicare are not covered by home infusion. Pharmacists should continue to try to persuade lawmakers to change this policy as this patient population would likely have great benefits in quality of life utilizing home infusion. Most home infusion services do have the option to see if coverage can be obtained through medical or prescription benefits. Pharmacies should review both options in order to get patients the most affordable care possible.
Another common site of care restriction is on growth factors. Often times our home infusion or specialty pharmacy can fill these and a patient can self-inject at home. For patients that are mandated to self-inject, the treatment nurse completes a teaching session with a syringe simulator on their first day of treatment. This ensures that the patient is competent to administer the medication in the home. The day after the dose is due, our home infusion pharmacy team calls to verify that the patient has administered the growth factor as a safety check.
Conclusions
Step edits and site of care restrictions are two major payer challenges in the oncology landscape. For step edits which are hard to stay on top off, implement a tracking plan to review payer policies at least twice a year if not quarterly and engage industry partners to help inform you of changes with their products. For site of care restrictions, get creative with how to keep these patients in-house and consider home infusion, self-injection, and utilizing your specialty pharmacy services.
Figure 1: Cancer Care and Home Infusion Collaboration Workflow
REFERENCES
- Modernizing Part D and Medicare Advantage To Lower Drug Prices and Reduce Out-of-Pocket Expenses. https://www.federalregister.gov/documents/2019/05/23/2019-10521/modernizing-part-d-and-medicare-advantage-to-lower-drug-prices-and-reduce-out-of-pocket-expenses. Accessed June 5, 2022.
Recipients of the Certificate of Recognition for Exemplary Research on Quality of Care in Oncology
Sol Atienza, PharmD, BCOP
Clinical Oncology Pharmacy Specialist
Advocate Aurora Health
Aurora St. Luke’s Medical Center
Milwaukee, WI
Michael Manlick, PharmD
Clinical Oncology Pharmacist
Advocate Aurora Health
Aurora St. Luke’s Medical Center
Milwaukee, WI
The Quality Oversight Committee of the Hematology/Oncology Pharmacy Association (HOPA) congratulate the recipients of the Certificate of Recognition for Exemplary Research on Quality of Care in Oncology. Although not everyone can be a recipient of the award, there is sincere gratitude to all who do research and, as a result, improve the care of our oncology patients. The submissions that were received underwent a review process for their respective categories. Each abstract was evaluated and scored based on set criteria specific to the care of patients with cancer. Quality, value metrics, and the potential impact on the current practice in oncology were all taken into consideration.
At the HOPA 2022 annual conference in Boston, MA, there were five certificate recipients of the abstracts submitted. The completed research certificate was received by Jodi Taraba, PharmD (Mayo Clinic and The University of Kansas Health System); and four trainee research certificates were awarded to Sara Bugamelli, PharmD (Henry Ford Hospital), Arielle Davidson, PharmD candidate 2024 (University of Michigan College of Pharmacy, Michigan Oncology Quality Consortium, and Michigan Institute for Care Management and Transformation), Sara Nowak, PharmD (St. Luke’s Hospital and University of Minnesota, Duluth College of Pharmacy), and Anastassia Blewett, PharmD (University of Virginia Health).
Development and Implementation of a Pharmacist-Led Virtual Clinic Improves Management of Metastatic Breast Cancer Patients on CDK 4/6 Inhibitors, presented by Jodi Taraba, PharmD
Managing patient care remotely, using a patient-centered model, can be a challenging practice. Taraba and colleagues developed a pharmacist-led multidisciplinary virtual clinic to improve the care of patients on cycle dependent kinase (CDK) 4/6 inhibitors. There is increasing use of CDK 4/6 inhibitors in combination with endocrine therapy for patients with advanced hormone receptor positive (HR+), human epidermal growth factor 2 (HER2) negative breast cancer. Patients receiving palbociclib, abemaciclib or ribociclib were managed by the virtual clinic. They were primarily contacted by phone every 2 weeks for the first two months and then monthly thereafter. These visits included review of lab results, toxicity assessment, medication adherence, medication reconciliation and screening for drug interactions. Laboratory adherence was compared against a retrospective cohort of patients initiated on palbociclib. Labs were appropriately drawn at 462/541 time points in the retrospective cohort (n=81) and at 128/139 time points in the prospective cohort (n=28), (85.4% vs 92.1%, p=0.038). Medication adherence to the prescribed medication was reported at > 99%. Interventions included patient education on administration of therapy thereby increasing adherence, symptom management, drug/supplement interactions and QT prolongation monitoring. There were also 38 potential drug-related issues identified. Improved care in patients receiving CDK 4/6 inhibitor therapy was demonstrated in this pharmacist-led clinic through improved laboratory adherence. With sustainment of this practice model, expansion to other oral therapies is being considered.
There are also future plans to develop and implement interactive care by utilizing technology to manage patients on CDK 4/6 inhibitors.
Evaluating the Impact of Prior Authorization on Oncology Patients Admitted to the Hospital, presented by Sara Bugamelli, PharmD
With increasing costs in cancer care, insurers have implemented requirements such as prior authorization (PA). Obtaining a PA can be a time intensive and complex process that can lead to treatment delays. Pharmacy involvement has been shown to decrease processing time, increase approval rates, and decrease incident of delayed discharges due to the PA process. Adding to the complexity with a PA-required medication, there is also the transition from the inpatient to outpatient setting. Objectives of this study were to identify predictive factors that identify the need for PA, to determine the impact of PA on inpatient length of stay, and to describe the process for assessing medication access prior to hospital discharge. Inpatient and outpatient pharmacy services collaborated to implement a standardized electronic test claim or Discharge Medication Cost Inquiry (DMCI) process to determine out-of-pocket cost of medications and the need for PA prior to hospital discharge. Bugamelli and colleagues conducted an IRB approved retrospective, comparative cohort study of patients admitted to the inpatient oncology unit between January 2019 and December 2020. Inclusion criteria were ≥ 18 years of age, admitted to the inpatient oncology unit for ≥ 72 hours, and had a documented DMCI for filgrastim, tbo-filgrastim, filgrastim-sndz, and/or filgrastim-aafi. Exclusion criteria were the vulnerable populations, patients enrolled in hospice or comfort care, those admitted with a diagnosis of acute myeloid leukemia, and patients admitted for stem cell transplant (SCT) or who have previously undergone SCT. The primary endpoint of the study was total length of hospital stay in days. Secondary endpoints include incidence of PA approval or denial, time to PA approval or denial in days, and delay in discharge defined as necessitating further overnight hospitalization once medically stable for discharge. Data collection included patient demographics, insurance type, reason for admission, biosimilar in DMCI documentation, and outcome of PA. Study results have shown (1) no significant difference in patient characteristics to anticipate the need for PA, (2) no significant difference in total hospital length of stay or delay in discharge in patients requiring a PA for GCSF, and (3) effective and efficient securing of PA’s by inpatient pharmacists prior to discharge through the DMCI process.
Standardized Outcome Measurement for Embedded Clinical Oncology Pharmacy Services, presented by Arielle Davidson, PharmD Candidate 2024
Oncology pharmacists continue to develop the role to strive to provide high-value and high-quality care to our cancer patients. Justifying the benefits of the oncology pharmacists remains difficult despite the many benefits that have been described in the literature. The roles of oncology pharmacists continue to evolve with increased integration into direct patient care and greater management of cancer therapy side effects and adherence. The main objective was to describe a model, Pharmacists Optimizing Oncology Care Excellence in Michigan (POEM), that supports the integration of clinical oncology pharmacists based in community practices while providing a standardized outcome assessment of practices to justify the pharmacist role. A database was developed to generate quarterly standardized reports. Patient surveys were also implemented to assess patient satisfaction. As of December 2021, POEM had 6 clinical oncology pharmacists representing 24 oncology clinics. To date, 1,424 patients had been seen via 2,893 encounters, which resulted in 3,065 pharmacist interventions. Demographics included a population that was 93% white, 5% black, 50% male, and 73% age equal to or greater than age 60. The primary reasons for pharmacist care were treatment with oral anticancer agents (OAA) (52%), non-immunotherapy IV (22%), immunotherapy (20%), and other (7%). A care management code was used to bill 66% of encounters. Types of interventions were education and referrals (52%), medication modification (24%), and comprehensive medication review or medication reconciliation (21%). Survey showed that patients felt it was important to meet with a pharmacist prior to beginning their cancer treatment. With success of justifying the beneficial role of the oncology pharmacist, this study supported expansion of the POEM program to 12 additional pharmacists to sites in 2022-2023. Future plans include analysis of pre- and post-data of healthcare utilization and patient risk.
The Impact of Patient and Provider Education on Colorectal Cancer Screening Rates at a Rural Clinic: A Quality Improvement Study, presented by Sara Nowak, PharmD
Guidelines for colorectal screening have recently been updated to help further identify the third most common cancer in the United States in men and women and leading cause of cancer-related death. Due to the growing incidence of colorectal cancer in younger patients, in 2021, the United States Preventative Services Task Force (USPSTF) expanded their screening recommendation to include patients aged 45-49 compared to their previous recommendation of beginning screening at the age of 50. Regular screening improves patient outcomes and reduces mortality. An initiative to achieve a screening rate of 80% in every community in the United States was established by the National Colorectal Cancer Roundtable. Pharmacists are well positioned to promote colorectal cancer screening through educational interventions. Nowak and colleagues conducted this quality improvement study to evaluate the impact of colorectal cancer screening rates through pharmacist provided education to patients and providers. The primary objective of the study was to assess post-intervention screening rates among previously unscreened adult patients aged 45 to 49 who receive care at a rural clinic. Secondary objectives include an assessment of the frequency of each type of screening (i.e., colonoscopy, stool tests, etc.), the number of abnormal findings through screening, the number of patients contacted who report they were already screened but were not identified in a generated report, and the perceived benefit of the provider education via a survey. The baseline screening rate was also determined. During the intervention period, patients were contacted via a mailed letter and scripted phone calls with education on colorectal cancer screening. Following the interventions, a chart review was conducted to determine the proportion of patients who have scheduled and completed colorectal cancer screening within a 3-month period. This study remains ongoing as patient interventions are further implemented; however, all providers felt that the live education increased their knowledge on colorectal screening. If further successful results, future plans are to expand to additional populations in the region.
Utilization of UVA Health Pharmacies for Outpatient Prescription Dispensation in the Malignant Hematology & Thoracic Oncology Clinics, presented by Anastassia Blewett, PharmD
An increase in prescription capture rates can lead to improved transitions of care, better medication adherence, and increased revenue for the organization. This retrospective chart review evaluated patients receiving prescriptions from the thoracic and malignant hematology clinics. The focus of the project was to implement interventions to improve prescription capture in their pharmacies and to determine how to assess and improve capture rates from outpatient oncology clinics. A multidisciplinary team evaluated the cancer center workflow and prescription filling process using a process mapping and fishbone diagram methodology. Between July 2020 to June 2021, the capture rate of the top oral non-chemotherapy prescriptions was 82.8%. The capture rate for the top oral chemotherapy and targeted agents from 2016 to 2021 was 44%. Barriers to prescription capture included drug availability, patient factors, pharmacy hours, staffing shortages, and lack of pharmacy advertisement. A Pareto chart illustrated the reasons for prescriptions not being filled. The most common reason being free drug, a close second reason was identified as contracts to outside specialty pharmacies, and the third reason was declining of treatment or transitioning to hospice. Future direction includes development of standardized reports to track prescription capture, discussing opportunities to negotiate insurance contracts, obtaining more resources to utilize pharmacy technicians, and advocate for increased staffing in the pharmacies to decrease dispensing delays and support medication delivery to patients.
Congratulations again to our award winners, and to everyone who presented their research at HOPA in 2022. All your hard work has not gone unnoticed and will bring change to our profession and, more importantly, improve patient care in the field of oncology.
Tebentafusp-tebn: Cytokine Release Syndrome Management and Operational Considerations
Ian Watson, PharmD
Clinical Oncology Pharmacist
M Health Fairview University of Minnesota Medical Center
Minneapolis, MN
Paul Morales, PharmD, BCOP
Pharmacy Manager
M Health Fairview Clinics and Surgery Center
Minneapolis, MN
Uveal Melanoma Treatment Landscape
Uveal melanoma, a distinct subset of melanoma, is the most common type of primary intraocular malignancy in adults.1 The reported incidence rates of uveal melanoma are 5.74 and 7.30 cases per million in North America and Europe respectively.2 Up to 50% of patients will have metastases, and the disease has a historically poor clinical response to systemic treatment.1,3 Meta-analyses combining results across studies of chemotherapy in metastatic uveal melanoma report median overall survival (OS) ranging from 9 to 11 months.1 Meta-analyses of anti-cytotoxic T-lymphocyte associated protein 4 (CTLA-4) therapy resulted in similar or worse outcomes compared to conventional chemotherapy.1 Mixed results have been seen with other immune checkpoint inhibitor regimens in this setting with positive results limited to phase 2 trials.1 The recent approval of tebentafusp-tebn (tebentafusp) by the United States Food and Drug Administration (FDA) offers the first treatment option specifically indicated for uveal melanoma. Tebentafusp is a bispecific Human Leukocyte Antigen (HLA)-A*02:01 directed CD3 T-cell engager.4 In the general uveal melanoma population, about 45% of patients are HLA-A*02:01-positive as determined by blood test.3 The drug has unique characteristics and a side effect profile that requires careful consideration when implemented into clinical practice.
Phase 3 Trial of Tebentafusp in Metastatic Uveal Melanoma3
A multicenter, randomized, phase 3 trial compared tebentafusp with investigator’s choice of single-agent pembrolizumab, ipilimumab, or dacarbazine as first-line systemic therapy in HLA-A*02:01-positive unresectable or metastatic uveal melanoma adult patients. The phase 3 trial included 378 patients assigned in a 2:1 ratio to tebentafusp or control. Tebentafusp was dosed once weekly with initial doses of 20 micrograms (mcg) on Week 1, 30 mcg on Week 2, followed 68 mcg weekly thereafter. The administration of tebentafusp resulted in a statistically significant improvement in median OS (21.7 months vs 16.0 months; Hazard Ratio [HR] for death 0.51, 95% Confidence Interval [CI] 0.37 – 0.71) and median progression-free survival (3.3 months vs 2.9 months; HR for disease progression or death 0.73, 95% CI 0.58 – 0.94).
Cytokine release syndrome (CRS) in the tebentafusp group was common, occurring in 89% of patients. The maximum grade of CRS was Grade 1 in 12% of patients, Grade 2 in 76%, Grade 3 in 1% (2 patients), and zero patients having Grade 4 or 5. Skin-related adverse events were also common and included rash (83%), pruritus (69%), and erythema (23%). This included a 21% incidence of Grade 3 skin reactions. No Grade 4 or 5 skin reactions or cases of Stevens-Johnson syndrome or toxic epidermal necrolysis were observed. The median time to onset of skin reactions was one day and most resolved to less than Grade 1 between tebentafusp doses. The incidence of these skin-related adverse events decreased with severity and frequency with subsequent infusions. Antihistamine and topical or systemic steroids were used to treat skin reactions based on persistence and severity of symptoms. No patients discontinued therapy due to skin reactions. Increases in alanine aminotransferase or aspartate aminotransferase occurred in 65% of patients with a 3% and 4% incidence of Grade 3 or higher respectively.
Tebentafusp-associated Cytokine Release Syndrome (CRS)
CRS is defined by the American Society for Transplantation and Cellular Therapy as “a supraphysiologic response following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells."5 Further, “symptoms can be progressive, must include fever at the onset, and may include hypotension, capillary leak (hypoxia) and end organ dysfunction.”5 CRS has been previously associated with chimeric antigen receptor (CAR) T-cell therapies and the only other FDA-approved bispecific T cell engager, blinatumomab.5
Since the approval of the first CAR T-cell therapy, there have been a few different methods used for CRS grading. The first five approved CAR T-cell therapies used the Lee 2014 criteria to grade CRS in their approval studies and thus use these criteria in their respective package inserts to align CRS grade with specific management recommendations.6-10 An exception is that in two of the first approval trials for tisagenlecleucel, the Penn criteria was used, which tend to assign a higher grade of CRS compared with the Lee 2014 criteria.5,6 The approval study and package insert for blinatumomab used the CTCAE version 4 criteria for CRS grading.12 In 2019, the American Society for Transplantation and Cellular Therapy (ASTCT) published consensus grading for CRS (Table 1).5 The new consensus grading has many similarities to the CTCAE criteria with the major differences being ASTCT specifically includes fever in the definition of CRS and is more specific with the types of interventions necessary in Grades 2-4.5 The ASTCT grading is also similar to the Lee 2014 criteria, however the former separates Grade 2 and 3 hypoxia by the device used to deliver oxygen (low-flow nasal cannula [≤6 L/minute] verse high-flow devices) instead of FiO2.5 Additionally, the ASTCT grading separates Grades 2-4 by the need and number of vasopressors required instead of the dose of vasopressor needed.5 Tebentafusp, as well as the most recently approved CAR T product, ciltacabtagene autoleucel, use the 2019 ASTCT consensus grading in their approval studies and package inserts.4,11
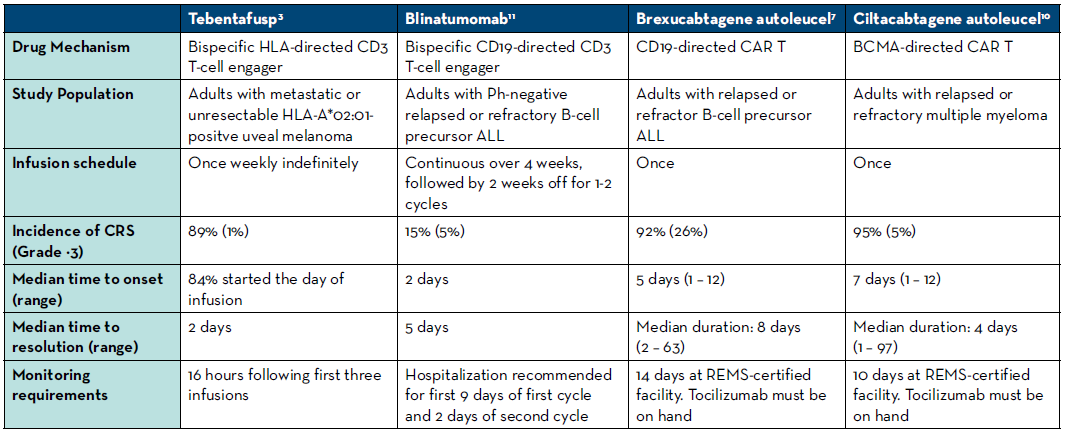
There are several important differences between the characteristics of CRS seen with tebentafusp compared to those seen with CAR T-cell therapies and blinatumomab. With tebentafusp, CRS occurred earlier (usually within a few hours after administration) and subsequently resolved earlier than with other therapies.4-12 Further characteristics of CRS are compared between a selection of these therapies in Table 2.
The treatment of CRS also varies for each therapy modality. Managing hypoxia and hypotension with oxygen supplementation and fluids or vasopressors is essential regardless of the immune therapy that caused CRS.5 Grade of CRS will need to be continually assessed based on interventions needed.5 Symptomatic treatment such as antipyretics may be beneficial for Grade 1.5 There are differences in when systemic steroids should be initiated for CRS treatment and what doses and formulations to use. Clinicians generally try to avoid corticosteroids until later in CRS associated with CAR T due to fear of causing death to the CAR T-cells and a subsequent reduction in efficacy. This risk is not a factor with the bispecific T-cell engager agents, and thus corticosteroids are often used earlier.4,12 Steroid doses and frequencies are escalated when CRS does not improve and when the grade increases.5 With tebentafusp, the manufacturer recommends administering an intravenous corticosteroid, such as methylprednisolone 2 mg/kg/ day or equivalent, beginning at Grade 3 CRS.4 The IL-6 receptor antagonist tocilizumab is an important part of CRS treatment with CAR T-cell therapies, but is not recommended for CRS of any grade associated with blinatumomab or tebentafusp.4-12 An additional consideration is the relatively short half-lives of blinatumomab and tebentafusp, which are 2.1 hours and 7.5 hours respectively. Stopping the infusion and/or delaying the next dose of these drugs can help minimize CRS duration – a strategy not available with one-time CAR T-cell infusions. Lastly, though distinct from CRS, neurotoxicity is a common and potentially severe adverse event associated with both CAR T-cell therapies and blinatumomab, but it has not been associated with tebentafusp.4-12
Operational considerations with tebentafusp
The CRS associated with tebentafusp warrants careful monitoring with appropriate tools in place to manage these events. In the Phase 3 trial, patients were monitored overnight after the first three infusions of the drug.3 The authors stated this was due to CRS events occurring in the hours after the first few doses.3 With the first three infusions of the drug the manufacturer recommends patients be monitored for 16 hours after administration, with vitals monitored at least every 4 hours.4 If a patient has not had hypotension requiring medical intervention with their most recent dose, a minimum of 30 minutes of observation after administration is required after week 4 and beyond with vitals measured twice post infusion.4 This presents a unique operational challenge as a patient will .need to receive at least their first 3 doses in a setting with nurse and provider access beyond the hours of a typical outpatient infusion center. Immediate access to emergency interventional tools including vasopressors, high flow oxygen, and mechanical ventilation with appropriately trained staff will also be necessary. The first 3 doses may need to be given in an outpatient clinic with extended hours or may require a one-night hospital admission. In this case, some institutions may be able to admit the patient after outpatient administration, under an observation status with different billing and reimbursement implications. Conversations with policy stakeholders, nursing staff, and provider groups will be important to find the optimal setting for health systems to administer tebentafusp.
Table 1. Summary of the ASTCT 2019 consensus CRS Grading5

Table 2. Comparison of CRS among selected bi-specific T cell engagers and CAR T-cell therapies
Also noteworthy is the preparation instructions for tebentafusp. To prevent adsorption of the drug to the infusion bag, human albumin must be added to a bag of 0.9% Sodium Chloride before the drug is added.4 Tebentafusp is supplied in a single-dose liquid preparation and the volume to be added to the prepared infusion bag is small, between 0.1 and 0.34 milliliters, depending on the dose.4 Following preparation, a 0.2 micron in-line filter infusion set primed with 0.9% Sodium Chloride must be added.4 The final product is stable for 4 hours at room temperature, and 24 hours refrigerated.4 Like other high-cost infusion products, the cost of the drug may limit the ability of institutions to prepare the product in advance of a patient’s appointment, as avoiding waste is important.
Conclusion
Tebentafusp offers a promising treatment option in the historically challenging to treat disease state of metastatic or unresectable uveal melanoma. While it is the second bispecific T cell engager to market, its exact mechanism is novel and the nature of its side effects are unique. Practitioners can borrow from their knowledge of CRS seen with CAR T-cell therapies and blinatumomab but must be aware of important differences with tebentafusp. The monitoring requirements of tebentafusp, particularly with the first three infusions, require careful planning to ensure an institution has the resources to safely treat uveal melanoma patients. With more immune therapies in the pipeline in various disease states, continuous discussions will be necessary to determine how to best administer these products.
REFERENCES
- National Comprehensive Cancer Network Guidelines 2.2022. Melanoma: Uveal. Updated April 5, 2022. Accessed June 1, 2022. https://www.nccn.org/professionals/physician_gls/pdf/uveal.pdf.
- Naseripoor M, Azimi F, Mirshahi R, Khakpoor G, Poorhosseingholi A, Chaibakhsh S. Global Incidence and Trend of Uveal Melanoma from 1943-2015: A Meta-Analysis. Asian Pac J Cancer Prev. 2022;23(5):1791-1801. doi:10.31557/APJCP.2022.23.5.1791.
- Nathan P, Hassel JC, Rutkowski P, et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N Engl J Med. 2021;385(13):1196-1206. doi:10.1056/NEJMoa2103485.
- Kimmtrak [package insert]. Conshohocken, PA: Immunocore Commercial LLC. 2022.
- Lee DW, Santomasso BD, Locke FL, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25(4):625-638. doi:10.1016/j.bbmt.2018.12.758.
- Kymriah [package insert]. Conshohocken, PA: Immunocore Commercial LLC. 2022.
- Yescarta [package insert]. Santa Monica, CA: Kite Pharma, Inc. 2022.
- Tecartus [package insert]. Santa Monica, CA: Kite Pharma, Inc. 2021.
- Breyanzi [package insert]. Bothell, WA: Juno Therapeutics, Inc., a Bristol- Myers Squibb Company. 2021.
- Abecma [package insert]. Summit, NJ: Celgene Corporation, a Bristol- Myers Squibb Company. 2021.
- Carvykti [package insert]. Horsham, PA: Janssen Biotech, Inc. 2022.
- Blincyto [package insert]. Thousand Oaks, CA: Amgen Inc. 2014.
Research Project Management
Katie S. Gatwood, PharmD, BCOP
Stem Cell Transplant & Cellular Therapy Clinical Pharmacist Specialist
PGY-2 Oncology Residency Program Director
Vanderbilt University Medical Center
Nashville, TN
The residency research project is one of the biggest hurdles residents face on the road to graduation. It is typically the most significant longitudinal requirement of the residency program and the one that most residents feel the most inexperienced with and therefore, can be especially daunting. As a residency program director and a regular research project preceptor, I will use this column to provide some tips and tricks to address some of the most common research project pitfalls.
Pitfall #1: Project selection
The project selection process can be highly variable among residency programs. Some programs provide residents with a list of project ideas to choose from and others will ask the resident to identify their own project. The latter can be especially challenging, particularly for residents who did not early commit to their program as they are also learning workflows and adapting to a new institution and practice site. It can also be more challenging for residents who have more limited prior experiences in oncology. It is hard to identify areas in need of clinical investigation when you are a novice in the field.
If you are in the position of having to identify your own project idea, I encourage you to utilize your preceptors and mentors to aid you in this process. Even something as simple as discussing topics you are interested in or questions you have regarding certain practices can end up sparking an idea. Also remember that just because you are in an oncology program does not mean your topic has to be strictly oncology focused. Any project that is conducted within an oncology population can inherently be considered an oncology project. Additionally, given the rapidity of new drug approvals in the oncology space, there is ample opportunity to study the many unknowns regarding proper use of these medications. Drug interactions and use in special populations or circumstances that were not included in clinical trials can be excellent research project ideas and there is always a need for more “real-world” data in the published literature.
For those residents who are selecting a project from a provided list, I believe there are some good things to keep in mind or ask of the project preceptor to ensure the highest likelihood of project success. First, consider the data sources for the project and how data will be obtained. For instance, do these sources require separate approval outside the IRB to obtain data or will they require any degree of manual review of a list of patients to identify those who can be include in the final data set? If so, these could be sources of potential delays. Another key consideration for project feasibility is the extent of manual chart review that is required. This is typically the most common issue I see leading to project delays so getting a realistic grasp on this before you begin the project is critical in ensuring it is a feasible project for the one-year timeline. If you can find a project that uses pre-existing data sources, such as an internal patient registry, or that largely includes data points that are discrete variables which can easily be pulled from the electronic medical record, I highly encourage selecting those. Finally, it is also important to determine the statistical plan for analysis of the results. Depending on how advanced the statistics needed are, you may need to plan for obtaining the help of a statistician or further training for yourself to conduct the analysis. Overall, I recommend selecting a project that is feasible, meaningful to practice, and of interest to you – in that order. Most graduating residents tell me that having a completed project that is easily publishable is more important than working on a project that they found to be the most interesting.
Pitfall #2: Project delays
Some sort of delay is almost inevitable with any research project, including residency research. There is always the potential for unforeseen circumstances that impact the project timeline. Therefore, I have two main tips to try and limit these and their impact as much as possible. First and foremost, make sure you thoughtfully consider the questions that I addressed in the project selection section above. This can help you predict where delays could occur so you can either prevent them or proactively identify strategies to deal with them. I encourage you to play a game of “worst case scenario” with your project preceptor where you try to identify all barriers you could encounter with the project. We do this within the Residency Research Advisory Committee at our institution prior to a resident submitting their project to the IRB and it frequently leads to changes to the original data collection plan or even the project proposal to help prevent common sources of delays, such as IRB revisions.
My other major piece of advice for dealing with project delays is to manage your time wisely and effectively. There is a lot of “down time” with research projects where you are waiting for something needed to move on to the next step, such as waiting for IRB approval, waiting to obtain a data set, or waiting for results to be analyzed. The biggest mistake you can make is to do no work on the project during these periods. If you use these periods to make some sort of forward progress, you will have more time available to manage and offset any delays that are likely to occur. For example, while waiting for IRB approval, you can work on your creating data collection tool, such as an Excel spreadsheet or a Redcap database. While waiting for your data set, you can create a plan with your preceptor on how you can get statistical assistance with the project if you know the analysis is beyond your capabilities, which could include securing funding for a statistician or taking an online biostatistics course. Finally, while waiting on your project results, you can begin to work on your abstract for submission to present as a poster at a professional meeting as well as work on the background and methods sections of a poster and/or manuscript so there is less to do when the data analysis is completed. Effective time management is one of the biggest keys to ensuring successful project completion when dealing with such a tight timeline.
Pitfall #3: Incomplete or unexpected results
Despite best efforts, there are still circumstances where residents do not have complete project results in time for presentation at a professional meeting or even for residency graduation. Alternatively, you may have results, but they are unexpected and dramatically alter the course of the project. Both scenarios can be incredibly stressful and disheartening when you have put such a great deal of time and effort into a project. Fortunately, all is not lost and there are still several ways that you can recover and make the most of what results you have.
First, you can still make an effective and meaningful presentation without complete results. Even if you don’t have any statistical analyses performed, you can often present demographic and qualitative data from the project that are still insightful. These data can tell a story, such as providing an overall incidence of an endpoint or outcome, even if you aren’t able to compare it to a control or historical group or link it to risk factors, as you may have planned in the original project proposal. If you have unexpected results, remember that these are still worth presenting and/or publishing. Trials with negative or “insignificant” results are infrequently published as they are often viewed as unimportant but there has been a push within the oncology community to publish more of these studies as they still provide valuable knowledge to apply to practice.
Finally, if you and your preceptor deem that your project truly cannot be published or presented due to incomplete or unexpected results, consider whether other projects you have worked on during the residency year can be published instead. In our program, we have had several residents who published the results of their medication use evaluations instead of their research projects. Otherwise, if obtaining a publication is one of your goals you can also talk to your program director and preceptors about other opportunities for this, such as writing a review article.
I hope these tips will prove useful to all of you out there embarking on the exciting journey of residency research. Remember that proper planning and some creative thinking can go a very long way in ensuring the success of your project!
Feature: Glucarpidase Dose Capping
Crystal Lu, PharmD, BCOP
Oncology Clinical Pharmacy Specialist
National Institutes of Health, Clinical Center Pharmacy Department
Bethesda, MD
The literature describing reduced-dose glucarpidase is limited to small patient populations, thus institutions and practitioners must weigh the risk versus the benefit of administering doses lower than the FDA-approved dose.
Too much of a good thing? The literature behind alternate glucarpidase dosing strategies.
Background
Methotrexate (MTX) is an antifolate chemotherapy agent that competitively inhibits the enzyme dihydrofolate reductase. It blocks the conversion of dihydrofolate to its active form, thus depleting the intracellular pool of tetrahydrofolates that are required as cofactors for the synthesis of thymidine and purines.1 MTX doses greater than 500mg/m2 are considered to be high dose (HD) and are indicated to treat adult and pediatric malignancies such as acute lymphoblastic leukemia, osteosarcoma, and lymphomas. High dose MTX-induced acute kidney injury (AKI) is an adverse event that may develop despite appropriate alkalinization, hydration, and leucovorin rescue. Since MTX is primarily eliminated through the kidneys, renal dysfunction delays MTX elimination and results in sustained elevated MTX plasma levels. This life-threatening toxicity puts patients at risk for MTX-related toxicities including myelosuppression, liver dysfunction, and mucositis.1-3 AKI is reported to occur in up to 12% of patients receiving HDMTX.4-7
Prior to the U.S. Food and Drug Administration (FDA) approval of glucarpidase, extracorporeal therapies were the primary modalities for removal of serum MTX. However, various methods of dialysis were not very effective due to the highly protein bound nature of MTX at 50% and the tendency for MTX levels to rebound.8-11 Historically, hemoperfusion/hemodialysis were used to remove serum MTX. The median serum MTX level decreases by 53% following the procedure, but later rebounds 20-90% of pre-dialysis levels. High-flux hemodialysis showed a median decrease of 75% of serum MTX levels and is now the most commonly used method of dialysis for MTX removal.5,12
Glucarpidase was approved in 2012 for treatment of toxic plasma MTX concentrations (>1 μmol/L) in adult and pediatric patients with delayed MTX clearance due to impaired renal function. Glucarpidase is a recombinant enzyme that hydrolyzes MTX and converts it to inactive metabolites 4-deoxy-4-amino-N10-methylpteroic acid (DAMPA) and glutamate. This allows for a non-renal pathway of elimination in patients with renal dysfunction.13 Consensus guidelines offer more stringent guidance for when to administer glucarpidase depending on the MTX dose, time from MTX infusion, and serum creatinine (Scr) increase from baseline.14 The Prescribing Information recommends glucarpidase dosing at 50 units/kg as a single intravenous injection.13 It is supplied as a lyophilized powder containing 1,000 units per vial. The approximate Average Wholesale Price (AWP) in 2022 is approximately $41,883 per vial.15 A 70 kg patient’s dose calculates to 3,500 units which will require four vials for compounding and cost $167,532. There is debate whether the manufacturer’s recommendation of 50 units/kg is the optimal dose since dose-finding studies were not performed in humans. More cost-effective approaches have been suggested which include administering less than 50 units/kg, capping the dose at a pre-specified number of vials, or rounding a patient’s dose to a whole vial. We summarize the literature using different glucarpidase dosing strategies in adult and pediatric populations.
Standard glucarpidase dosing in adults and pediatrics
The efficacy of glucarpidase was studied in a subset of 22 patients (12 pediatric and 10 adults) in a single-arm, open-label study. Patients had renal dysfunction and delayed MTX clearance defined as >2 standard deviations greater than the mean excretion curve. Glucarpidase was dosed at 50 units/kg and a second dose was given to patients with pre-glucarpidase MTX concentrations >100 μmol/L. The main outcome measure was the proportion of patients who achieved a rapid and sustained clinically important reduction (RSCIR) in plasma MTX concentration, defined as plasma MTX <1 μmol/L at 15 minutes and sustained up to 8 days. Ten patients (45%) achieved RSCIR and 5 patients (23%) attained a transient plasma MTX concentration <1 μmol/L. These results led to the full FDA approval of glucarpidase in 2012 for delayed MTX clearance due to HDMTX induced AKI.13
Alternate dosing strategies in adults
In a multicenter study, Schwartz and colleagues evaluated glucarpidase intervention in adult and elderly patients with delayed MTX elimination. Forty-three patients received the standard 50 units/ kg dose and a second dose was optional in those with serum MTX concentrations >0.1 μmol/L at >24 hours after the first intervention. The median age was 54 years (range 18-78). Investigators saw a >97% reduction in serum MTX by high-performance liquid chromatography (HPLC). Of note, a subset of 11 patients received lower glucarpidase doses ranging from 10 to 31 units/kg due to a transient shortage of supply. Normalization or improvement in SCr was seen in 93% (40/43) of patients. Serum samples for immunogenicity were collected for 7 patients. Three of the 7 patients developed antiglucarpidase antibodies between 7-17 days after glucarpidase administration. Only one of the antibody-positive samples showed a slight reduction (23%) in glucarpidase activity. Detailed efficacy in the 11 patients who received the lower doses were not provided. Grade 3 and 4 toxicities, graded according to the World Health Organization (WHO), include hematological (60%), mucositis (35%), renal (19%), liver (16%), central nervous system (14%), and skin (2%).16
Heuschkel and colleagues evaluated half-dose glucarpidase at 25 units/kg in 7 adults with toxic MTX plasma concentrations. Patients ranging from 19 to 71 years of age received glucarpidase a median of 58 hours (range 42-72 hours) after the initiation of MTX. Within one day of glucarpidase administration, the MTX plasma concentrations decreased by 97.5%. The authors noted methotrexate rebound 42-73 hours after methotrexate initiation; however, the MTX concentration was consistently less than 10 μmol/L. Of the 6 patients with initial evaluable levels, five patients (83%) had MTX concentrations <1 μmol/L. All patients had levels that remained permanently <10 μmol/L. One patient received intermittent veno-venous hemodialysis because of a very high MTX level and delay in glucarpidase delivery. Significant toxicities > Grade 3 per Common Terminology Criteria for Adverse Events (CTCAE) include hepatotoxicity in 1 patient (14%), thrombocytopenia in 3 patients (43%), and leukopenia in 2 patients (29%).17
Several case reports and case series describe other methods for glucarpidase dosing. Trifilio et al. describes a case of a 59-year-old man with newly diagnosed pre-T acute lymphoblastic lymphoma starting part B of hyper-CVAD. After an increase of Scr to 2.58 mg/dL at 24 hours post-MTX and 4.54 mg/dL at 48 hours post-MTX dose, the patient became anuric. After two sessions of high-flux hemodialysis, the patient was treated with a single 1,000 unit dose of glucarpidase, which was approximately 15% of the approved dose. The MTX level 12-hours post-glucarpidase was 0.5 μmol/L. His MTX levels continued to decline and SCr improved over the next 5 days.18 In a case series by Krüger et al., the authors describe 3 obese patients with elevated levels 24 hours post-MTX at 97, 52, and 19 μmol/L. Of the 3 patients, two experienced AKI. After a single dose of glucarpidase 4,000 units (approximately 50 units/kg per ideal body weight), MTX levels quickly dropped to 1.37, 0.07, and 0.03 μmol/L, respectively.19 A recent case series described 5 patients who received HDMTX infusions and had elevated 48-hour MTX levels >5 μmol/L and SCr increase >50% from baseline. The adults ranged from 38-74 years and weighed 69-156 kg. A fixed dose of 1,000 units of glucarpidase was given and all 5 patients had MTX levels undetectable within 24 hours of glucarpidase administration.20
Alternate dosing strategies in pediatrics
Scott et al. retrospectively evaluated 26 pediatric patients ranging 4.0- 20.4 years of age who received HDMTX at St. Jude Children’s Research Hospital. Methotrexate doses were rounded to the nearest vial size and doses ranged from 13 to 90 units/kg. Patients who received glucarpidase <50 units/kg had median percent reduction in plasma concentration of 99.4% (range 90-100%) measured by HPLC compared to median percent reduction of 99.4% (range 77.2-100%) in patients who received >50 units/kg. The authors concluded there was no significant relationship between the glucarpidase dose and percent decrease in methotrexate plasma concentrations. There was also no statistically significant association found between glucarpidase dose and time to SCr recovery (P>0.8).21
Other considerations
One of the challenges in evaluating the efficacy of glucarpidase is the institution’s ability to accurately measure serum MTX levels. Within 48 hours after glucarpidase administration, immunoassays are unreliable and will overestimate the MTX level.13 The metabolite DAMPA cross reacts with serum MTX that is measured with the standard immunoassay. HPLC is a more sensitive method that can accurately measure serum MTX levels even during the 48-72 hour period after glucarpidase administration; however, its availability may vary across hospitals.14 One study suggests that it may take even longer than 72 hours for immunoassays to not overestimate MTX levels, thus making it challenging for providers practicing at hospitals without HPLC to know if a post-glucarpidase MTX level is accurate or possibly falsely elevated.22
The literature describing reduced-dose glucarpidase is limited to small patient populations, thus institutions and practitioners must weigh the risk versus the benefit of administering doses lower than the FDA-approved dose. A major driving factor for using a lower dose is the high cost for each vial. Importantly, lower doses can also be justified pharmacokinetically, since glucarpidase has a mean volume of distribution of 3.6 L suggesting its distribution is restricted to plasma volume.13 Glucarpidase reduces MTX concentrations in the intravascular compartment; however, it has no impact on MTX located intracellularly or within the urinary collecting system. Thus, leucovorin rescue is still needed to address the intracellular compartment.23 If institutions decide to routinely give a flat dose such as 2,000 units (2 vials), they may be able to justify carrying a supply on-site at all times to ensure timely administration. Some may argue that if lower doses are given initially, a second dose may be given at a later time point, if necessary. However, Widemann et al. reported that subsequent second and third doses of glucarpidase did not substantially decrease plasma MTX concentrations.7 Theoretically, a second dose may not be as effective in preventing MTX-induced toxicities, since by this time the majority of MTX may already be located intracellularly or converted to inactive metabolites with the first glucarpidase dose. Another limitation to subsequent doses is the potential development of antibodies to glucarpidase.
The management of toxic MTX concentrations is undoubtedly a high cost to the healthcare system. However, administration of the FDA-approved dose of glucarpidase may be the most quick and effective method proven to decrease serum MTX levels. Some may argue that perhaps it may be better to give the full dose rather than risking an inadequate decrease in MTX levels and potentially acquire other indirect costs such as increased length of intensive care unit stay or dialysis. Renal replacement therapy in the form of hemodialysis or hemoperfusion may be used to remove MTX levels from the blood. However, the decline in MTX levels is slow with frequent rebound.7 A retrospective study in patients admitted for HDMTX who subsequently developed MTX toxicity found that patients treated with glucarpidase had shorter average length of stay of 14.7 days compared to all nonglucarpidase patients at 21.9 days and nonglucarpidase patients who received dialysis of 40.2 days.24 The study was not designed to compare length of stay among patients receiving different glucarpidase doses. It is important to also note that multiple dialysis sessions increase the risk for infection, bleeding complications, thrombocytopenia and electrolyte disturbances.
If debating whether glucarpidase is indicated in a unique patient scenario, a set of consensus guidelines offers roadmaps for decision making.14 Additionally, an open-access clinical decision-making tool is available at MTXPK.org. It uses patient-specific characteristics and pharmacokinetic modeling to compare expected vs. actual MTX elimination time curves.25,26 If glucarpidase is needed, distributors are able to send drug to hospitals through expedited or next day delivery.27 Administration of drug is time-sensitive, as early administration <96 hours after HDMTX exposure has been shown to decrease MTX-related toxicities. Grade 4 toxicity developed in 9/64 (14%) patients who received glucarpidase <96 hours compared to 6/11 (55%) of patients >96 hours.28 The availability of drug and treatment timeline should be considered when evaluating whether to keep drug on-hand. Currently, practices vary by hospital. For example, the Mayo Clinic implemented an institutional protocol to administer fixed-dose glucarpidase at 1,000 units/dose and to carry 2 vials on-site after evaluating successful results in their case series.20 The consensus guidelines for stocking antidotes recommend routinely stocking 5 vials.29
Conclusion
Because glucarpidase is an antidote, its rare indication and limited number of patients in studies makes it challenging to determine the most effective, safe, and economical dosing strategy. It is the responsibility of each institution that administers HDMTX to assess its need to add glucarpidase to formulary and determine how to routinely stock or procure the medication.
REFERENCES
- Widemann BC, Adamson PC. Understanding and managing methotrexate nephrotoxicity. Oncologist. 2006;11:694-703.
- Abelson HT, Fosburg MT, Beardsley GP, et al. Methotrexate-induced renal impairment: Clinical studies and rescue from systemic toxicity with high-dose leucovorin and thymidine. J Clin Oncol. 1983;1(3):208-216.
- Relling MV, Fairclough D, Ayers D, et al. Patient characteristics associated with high-dose methotrexate concentrations and toxicity. J Clin Oncol. 1994;12(8):1667-1672.
- Hoang-Xuan K, Taillandier L, Chinot O, et al. Chemotherapy alone as initial treatment for primary CNS lymphoma in patients older than 60 years: A multicenter phase II study (26952) of the European Organization for Research and Treatment of Cancer Brain Tumor Group. J Clin Oncol. 2003;21(14):2726-2731.
- Widemann BC, Balis FM, Kempf-Bielack B, et al. High-dose methotrexate-induced nephrotoxicity in patients with osteosarcoma. Cancer. 2004;100:2222-2232.
- Jahnke K, Korfel A, Martus P, et al. High-dose methotrexate toxicity in elderly patients with primary central nervous system lymphoma. Ann Oncol. 2005;16(3):445-449.
- Widemann BC, Balis FM, Kim A, et al. Glucarpidase, leucovorin, and thymidine for high-dose methotrexate-induced renal dysfunction: Clinical and pharmacologic factors affecting outcome. J Clin Oncol. 2010;28(25):3979-3986.
- Wall SM, Johansen MJ, Molony DA, et al. Effective clearance of methotrexate using high-flux hemodialysis membranes. Am J Kidney Dis. 1996;28(6):846-854.
- Diskin CJ, Stokes TJ, Dansby LM, et al. Removal of methotrexate by peritoneal dialysis and hemodialysis in a single patient with end-stage renal disease. Am J Med Sci. 2006;332(3):156-158.
- Vilay AM, Mueller BA, Haines H, et al. Treatment of methotrexate intoxication with various modalities of continuous extracorporeal therapy and glucarpidase. Pharmacotherapy. 2010;30(1):53e-58e.
- Connors NJ, Sise ME, Nelson LS, et al. Methotrexate toxicity treated with continuous venovenous hemofiltration, leucovorin and glucarpidase. Clin Kidney J. 2014;7(6):590-592.
- Kitchlu A, Shirali AC. High-flux hemodialysis versus glucarpidase for methotrexate-associated acute kidney injury: What’s best? J Onco Nephrol. 2019;3(1):11-18.
- Voraxaze; [package insert]. BTG International Inc; Brentwood, TN: 2012.
- Ramsey LB, Balis FM, O’Brien MM, et al. Consensus guideline for use of glucarpidase in patients with high-dose methotrexate induced acute kidney injury and delayed methotrexate clearance. Oncologist. 2018;23:52-61.
- Glucarpidase. Lexi-Drugs. Hudson, OH: Lexicomp, 2022. http://online.lexi.com/. Updated May 10,2022. Accessed June 29, 2022.
- Schwartz S, Borner K, Müller K, et al. Glucarpidase (Carboxypeptidase G2) intervention in adult and elderly cancer patients with renal dysfunction and delayed methotrexate elimination after high-dose methotrexate therapy. Oncologist. 2007;12:1299-1308.
- Heuschkel S, Kretschmann T, Teipel R, et al. Half-dose glucarpidase as efficient rescue for toxic methotrexate levels in patients with acute kidney injury. Cancer Chemother Pharmacol. 2022;89:41-48.
- Trifilio S, Ma S, Petrich A. Reduced-dose carboxypeptidase-G2 successfully lowers elevated methotrexate levels in an adult with acute methotrexate-induced renal failure. Clin Adv Hematol Oncol. 2013;11(5):322-325.
- Krüger C, Engel N, Reinert J, et al. Successful treatment of delayed methotrexate clearance using glucarpidase dosed on ideal body weight in obese patients. Pharmacotherapy. 2020;40(5):479-483.
- Truong H, Leung N. Fixed-dose glucarpidase for toxic methotrexate levels and acute kidney injury in adult lymphoma patients: Case series. Clin Lymphoma Myeloma Leuk. 2021;21(6):497-502.
- Scott JR, Zhou Y, Cheng C, et al. Comparable efficacy with varying dosages of glucarpidase in pediatric oncology patients. Pediatr Blood Cancer. 2015;62(9):1518-1522.
- Gulley SL, Franco KA, Elder JJ. Monitoring methotrexate levels in a patient after glucarpidase administration: A case report. J Hematol Oncol Pharm. 2020;10(3):154-157.
- Meyers PA. Glucarpidase for the treatment of methotrexate-induced renal dysfunction and delayed methotrexate excretion. [Letter to the editor]. Pediatr Blood Cancer. 2016;63:364.
- Demiralp B, Koenig L, Kala J, et al. Length of stay, mortality, and readmissions among Medicare cancer patients treated with glucarpidase and conventional care: a retrospective study. Clin Outcomes Res. 2019;11:129-44.
- MTXPK.org. https://mtxpk.org/. Accessed 14 Jun 2022.
- Taylor AL, Mizuno T, Punt NC, et al. MTXPK.org: a clinical decision support tool evaluating high-dose methotrexate pharmacokinetics to inform post-infusion care and use of glucarpidase. Clin Pharm Therap. 2020;108(3):635- 643.
- VORAXAZE (glucarpidase). https://voraxaze.com/. Accessed 14 Jun 2022.
- 2013 Annual Meeting of the North American Congress of Clinical Toxicology (NACCT). Clin Toxicol. 2013;51(7):575-724.
- Dart RC, Goldfrank LR, Erstad BL, et al. Expert consensus guidelines for stocking antidotes in hospitals that provide emergency care. Ann Emerg Med. 2018;71(3):314-325.
Pharmacists and Patient Advocacy Organizations Working Together to Fill the Gaps in Care
George Valentine
Patient Advisory Panelist for HOPA
Retired Information Technology Consultant
Irving, TX
Chelsea Gustafson, PharmD, BCOP
Oncology Clinical Pharmacy Specialist
Community Health Network MD Anderson Cancer Center
Kokomo, IN
At this year’s annual conference, the Patient Outreach Committee hosted a session that brought together advocacy organization representatives, HOPA members, and Patient Advisory Panelists. The session allowed for small groups to meet and discuss the offerings of the represented organizations and learn how they could use the resources from these organizations to support patients. Patient Advocate Foundation (PAF), Stupid Cancer, Fight Colorectal Cancer (Fight CRC), and St. Baldrick’s Foundation were all represented at the session.
Participants learned about Fight CRC’s advocacy on the hill efforts and a multitude of videos available online for patients and caregivers to hear firsthand from other patients about their experiences in their cancer journey. They heard how Stupid Cancer offers a lifeline to adolescent and young adults (AYA) with cancer by connecting them with resources and peers and identifying the unique challenges of cancer in that stage of life. Representatives from St. Baldrick’s Foundation shared ways that pharmacists can get involved by volunteering, fundraising, and advocating for key legislation that impacts kids with cancer. Attendees also heard about some of the many things that the Patient Advocate Foundation (PAF) does to provide case management services and financial aid to people with not only cancer, but many chronic, life-threating, and debilitating illnesses. These were just some of the highlights of what was discussed.
George Valentine is a panelist on HOPA’s Patient Advisory Panel. He attended HOPA’s Annual Conference this year and participated in the advocacy session, Pharmacists and Patient Advocacy Organizations Working Together to Fill the Gaps in Care. He answered some questions and provided some of his takeaways from the session.
What services provided by the advocacy organizations do you think will be most impactful for pharmacists to share with individual patients?
While most advocacy organizations have a common goal to improve life for people living with cancer, their services and activities vary from one to the next. People living with cancer deal with a wide range of challenges including treatment options, choosing the right care facility and doctor, drug cost, work and or family responsibilities, access to information, finding support groups and finding a means to express their concerns. Every pharmacist must understand that two patients taking the same drug will not likely share the same set of challenges. Advocacy organizations can be a critical information resource to all pharmacists treating cancer patients.
What were you surprised to learn about during the session that you would like to share?
I was surprised to learn that there are so many different advocacy organizations of which I as a 20-year cancer survivor never knew existed. I left the session with mindset that access to needed healthcare with affordable treatment and drug cost is still a long way off and without the constant push by advocacy organizations we would continue to see needless adverse patient outcomes.
What initiatives did you learn about that you think will help improve care for cancer patients in the future?
I learned of efforts to influence/change our governmental policies that influence or direct every aspect of my healthcare as a cancer patient. As a retired senior on Medicare these initiatives would be most impactful to me as Medicare has cumbersome treatment guidelines and no existing limit/cap on out-of-pocket prescription drug cost.
How have advocacy organizations impacted your own personal cancer journey?
Several years ago, the doctor treating my Chronic Lymphocytic Leukemia (CLL) moved me from in-hospital treatment to an oral cancer drug taken daily. The drug improved every aspect of my treatment and life, but its cost was over $12,000 dollars a month with a high out-of-pocket burden. My pharmacists suggested I reach out to the Patient Access Network (PAN). The PAN Foundation provided me with financial assistance which covered 100% of my out-of-pocket for that specific drug. I do not feel I would be alive today were it not for the support of PAN.
PAN also has formal ongoing efforts advocating for improved drug access and affordability. PAN has submitted over fifteen policy recommendations, seventeen informational briefs, and thirteen policy letters to congress.
What is something new that you learned about the role of oncology pharmacists?
I live in a large city with options for hospitals and pharmacies. I was unaware of the expansive services and support provided by pharmacists who operate in remote/rural areas. I was also impressed by the amount of continuing education required and completed by oncology pharmacists.
What were your overall thoughts of attending HOPA AC 2022?
Having had experiences with a limited number of pharmacists, I was surprised by the diversity of attendees at the conference. I met and spoke with people from various cultures, locations, and educational backgrounds. I, like most cancer patients, see diversity as a positive option when choosing healthcare providers. I gained a deeper understanding of the role HOPA plays in bolstering the competencies of oncology pharmacists. I really enjoyed the poster board sessions that allowed me to see the planned improvements by prescription drug manufacturers and the availability of clinical trials. My heartfelt thanks to the team of people who put together an experience I will always remember.
After attending the conference, do you feel your role or thoughts on being a panelist have changed? If so, how?
I attended the conference with a feeling that advocacy could be a means for me to give back to all the people who have cared for me over the years. The conference energized me to take the next step to create an advocacy goal and plan of action for the next 12 months. I look forward to my continued participation as a panelist.
We thank George and all of our Patient Advisory Panelists for their participation in the advocacy session and HOPA AC22.
Evaluation of Clostridioides Difficile Infection (CDI) Treatment Duration in Hematology/Oncology Patients Receiving Other Concurrent Antibiotics
Amber B. Clemmons, PharmD, BCOP, FHOPA
Clinical Professor, University of Georgia College of Pharmacy
Clinical Pharmacy Specialist
Augusta University (AU) Medical Center
Augusta, GA
Tia Stitt, PharmD
Clinical Infusion Pharmacist
Georgia Cancer Center at the AU Medical Center
Augusta, GA
Clostridioides difficile infection (CDI) is a nosocomial infection with symptoms ranging from mild to severe, including fulminant disease with toxic megacolon. Hematology/oncology patients are at higher risk for developing CDI compared to the general population (6-33% versus 1-2%).1-4 These patients are immunocompromised with an increased susceptibility to infectious processes and are frequently placed on broad-spectrum non-CDI antibiotics for prevention and treatment of bacterial infections. These agents vary in risk of developing CDI, with fluoroquinolones, cephalosporins, carbapenems, and beta-lactam/beta-lactamase inhibitors being considered high risk.5-6 Notably, these agents are frequently utilized in this patient population. Further, hematology/oncology patients are at higher risk of recurrent infections (20-40% versus 20%) which are associated with higher healthcare costs and lower quality of life.4, 7-8
Currently, the Infectious Disease Society of America (IDSA) guideline recommends treating CDI for 10-14 days.9 Additional recommended interventions include hydration and electrolyte repletion, infection control measures such as adequate handwashing, and discontinuation of non-CDI antimicrobials as soon as possible. However, the latter recommendation is challenging to implement for hematology/oncology patients given their immunocompromised state and frequent need for non-CDI antimicrobials to prevent and manage bacterial infections.
Further interventions to prevent recurrence of CDI are of much interest. Specifically, the use of CDI targeted agents has been evaluated, although only minimally in the hematology/oncology population. Prophylactic CDI treatment in patients with a history of CDI and receiving non-CDI antibiotics has been shown to be effective in a few studies.10-12 However, limited data exists regarding the extension of CDI treatment for those without history of CDI who are on concurrent non-CDI antibiotics. Due to the limited data, the IDSA guideline states that insufficient data exists to recommend extending CDI treatment beyond the recommended duration even in those patients receiving concurrent non-CDI antibiotics.9 Although minimal data is available, some providers at our institution have used prolonged or extended duration of therapy for patient on concurrent non-CDI antibiotics to prevent CDI recurrence.
To further address the impact of prolonged or extended CDI treatment in hematology/oncology patients on non-CDI antibiotics, particularly on recurrence of CDI, a joint PGY1 pharmacy residency research project was performed by Stitt and colleagues at the Phoebe Putney Memorial Hospital in Albany, Georgia along with Keats and colleagues at the AU Medical Center in Augusta, Georgia.13
This study was a multi-site, retrospective evaluation of hematology/oncology patients aged 18 years and older who were admitted to either facility between September 2013 – June 2019 with an active CDI diagnosis who received at least one dose of concurrent non-CDI antibiotics within 24 hours of CDI diagnosis or at any time during CDI treatment. Patients were classified by two different definitions for evaluation of outcomes. In the first analysis, patients were divided into cohorts by duration of CDI treatment: standard (10-14 days) versus prolonged (> 14 days). In the second planned analysis, the same patients were divided into cohorts by CDI treatment duration in relation to the end of non-CDI antibiotic treatment: non-extended (CDI treatment ended ≤ 24 hours after stopping non-CDI antibiotic) versus extended (CDI treatment ended > 24 hours after stopping non-CDI antibiotic). In both analyses, the primary outcome was incidence of CDI recurrence within 180 days of completing CDI treatment. Secondary outcomes included hospital length of stay (LOS), incidence of vancomycin-resistant enterococcus (VRE) infections at 180 days, and mortality rate at 180 days.
Table 1: Key Findings
One hundred and ninety-eight patients were included in the analysis; of these, 112 were classified as prolonged duration versus 86 standard duration for the first analysis, as well as 138 classified as extended duration versus 60 non-extended duration for the second analysis. Both sets of cohorts were well balanced with respect to history of previous CDI episode, severity of CDI on diagnosis, and receipt of proton pump inhibitor. Differences in diagnosis were observed within the prolonged versus standard cohort. More patients with hematologic malignancies were included in the prolonged group and more patients with solid tumors were included in the standard group. With respect to the primary outcome, the incidence of CDI recurrence was not different between either the standard versus prolonged or between the extended versus non-extended duration cohorts (all p > 0.05). For the standard versus prolonged analysis, hospital length of stay was longer in prolonged cohort (27 vs 12 days; p<0.0001) but there was no difference in mortality (p > 0.05). For the non-extended versus extended analysis, no difference existed in hospital LOS and the mortality was lower in extended cohort (p=0.0008). Overall, a low incidence of VRE infection was documented which did not differ between standard versus prolonged cohorts (3% vs 8%; p>0.05) or between non-extended versus extended cohorts (8% vs 5%; p>0.05).
Limitations of our study include low number of patients in one of the two sites, retrospective and non-randomized nature of the study design, and inability to obtain data from outside our institutions. Ergo, under-reporting of outcomes such as CDI recurrence and death could have occurred. Further, inability to evaluate data by sub-groups, such as specific type of cancer or transplant, hinders interpretation as certain groups may be higher risk and be more likely to benefit from longer duration. Hence, further prospective and controlled trials would provide guidance to those trying to manage CDI treatment in this higher risk population. These studies should evaluate patients receiving high versus lower risk antibiotics separately, which was not able to be captured in our study due to low incidence of low-risk antibiotic use in our population. Further, future studies should evaluate specific populations at highest risk for CDI recurrence such as those with multiple risk factors or prior CDI episodes.
Providers should carefully consider the aforementioned findings before prolonging or extending the duration of CDI treatment. Given a dearth of supportive data, this practice should not be advised routinely at this time.
REFERENCES
- Kamthan AG, Bruckner HW, Hirschman SZ, et al. Clostridium difficile diarrhea induced by cancer chemotherapy. Arch Intern Med 1992; 152: 1715–1717.
- Husain A, Aptaker L, Spriggs DR, et al. Gastrointestinal toxicity and Clostridium difficile diarrhea in patients treated with paclitaxel-containing chemotherapy regimens. Gynecol Oncol 1998; 71: 104–107.
- Khan A, Raza S, Batul SA, et al. The evolution of Clostridium difficile infection in cancer patients: epidemiology, pathophysiology, and guidelines for prevention and management. Recent Pat Antiinfect Drug Discov 2012; 7: 157–170.
- Revolinski SL and Munoz-Price LS. Clostridium difficile in immunocompromised hosts: a review of epidemiology, risk factors, treatment, and prevention. Clin Infect Dis off Dis 2019; 68: 2144–2153.
- Deshpande A, Pasupuleti V, Thota P, et al. Community associated Clostridium difficile infection and antibiotics: a meta-analysis. J Antimicrob Chemother 2013; 68: 1951–1961.
- Brown KA, Khanafer N, Daneman N, et al. Meta-analysis of antibiotics and the risk of community-associated Clostridium difficile infection. Antimicrob Agents Chemother 2013; 57: 2326–2332.
- Chung MS, Kim J, Kang JO, et al. Impact of malignancy on Clostridium difficile infection. Eur J Clin Microbiol Infect Dis 2016; 35: 1771–1776.
- Madoff SE, Urquiaga M, Alonso CD, et al. Prevention of recurrent Clostridioides difficile infection: A systematic review of randomized controlled trials. Anaerobe 2020; 61:102098.
- McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis off Dis 2018; 66: e1–e48.
- Ganetsky A, Han JH, Hughes ME, et al. Oral vancomycin prophylaxis is highly effective in preventing Clostridium difficile infection in allogeneic hematopoietic cell transplant recipients. Clin Infect Dis off Publ Infect Dis Soc Am 2019; 68: 2003–2009.
- Van Hise NW, Bryant AM, Hennessey EK, et al. Efficacy of oral vancomycin in preventing recurrent Clostridium difficile infection in patients treated with systemic antimicrobial agents. Clin Infect Dis 2016; 63: 651–653.
- Carignan A, Poulin S, Martin P, et al. Efficacy of secondary prophylaxis with vancomycin for preventing recurrent Clostridium difficile infections. Am J Gastroenterol 2016; 111: 1834–1840.
- Keats K, Stitt T, Chastain D, Jivan B, Matznick E, Waller J, Clemmons AB. Evaluating Clostridioides difficile infection (CDI) treatment duration in hematology/oncology patients receiving concurrent non-CDI antibiotics. J Oncol Pharm Pract 2021 DOI: 10.1177/1078155221998735.
Tebentafusp (Kimmtrak)
Aaron C. Johnson, PharmD candidate 2023
University of Michigan, College of Pharmacy
Ann Arbor, MI
Shawna Kraft, PharmD, BCOP
University of Michigan, College of Pharmacy
University of Michigan, Rogel Cancer Center
Ann Arbor, MI
Background Uveal Melanoma
Uveal melanoma (UM) is an especially rare cancer, occurring in 1 per 200,000 Americans per year.1 While it shares a name with cutaneous melanoma, it is its own distinct entity. Uveal melanomas have specific biomarkers, stemming from oncogenic mutations, that set it apart from the cutaneous or mucosal melanomas. UM prognosis is dependent on disease severity; patients with localized disease have an 85% or better probability of survival 5 years from diagnosis, whereas those with metastatic disease have a less 20% probability of being alive at 5 years.2-4 The lack of reliability amongst current treatments for UM presents a unique opportunity to engage innovative therapies for the treatment of this aggressive and difficult to treat cancer.
UM arises from the melanocytes located in the uveal tract of the eye. UM tumors rarely reveal metastasis at the primary diagnosis, but 50% of patients develop metastasis despite eradication of the primary tumor. This correlation poses the possibility of dormant micrometastases that remain undetected for many years.4
The low mutation burden of UM, less than one single nucleotide variation (SNP), is far lower than many other cancer types.4 Two proposed mutations of UM are guanine nucleotide-binding protein alpha Q (GNAQ) and guanine nucleotide-binding protein alpha (GNA11). Each of these mutations regulate the exchange of GDP for GTP which regulates the growth, differentiation, and migration of tumors, however, they are not directly correlated to the rates of metastasis. The deubiquitinating of proteins is a key step in the regulation of cellular proliferation. Ubiquitin carboxyl-terminal hydrolase (BAP1) is a deubiquitinating enzyme linked to having a high presence in multiple domains of UM tumors.5,6 Functioning as a tumor suppressor gene, BAP1 has a plethora of downstream functions directly linked to the maintenance of the cell cycle, but its ultimate role on metastasis of UM is still unknown.6
Effective treatments are lacking for metastatic UM. Systemic treatments from previously reported trials include intravenous chemotherapy such as dacarbazine, cisplatin or nitrosourea, all having modest impact on overall survival.5 Ipilimumab is approved in the U.S. for treatment of unresectable melanoma, however, with response rates less than 10%, the data reflects a slow and delayed response to a minority of metastatic UM patients.2 Thus until recently, the National Comprehensive Cancer Network (NCCN) recommended pursuit of a clinical trial as front line therapy for metastatic UM, with liver-directed therapies, radiation strategies, or best supportive care as alternatives.7
Tebentafusp-tebn Characteristics
Tebentafusp-tebn is the first FDA T-cell receptor therapy approved for HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma.9 Tebentafusp-tebn is a bispecific protein composed of a modified T-cell receptor fused to an anti-CD3 single chain fragment. This medication is classified as an immune-mobilizing T-cell receptors against cancer (ImmTAC) which are proteins specifically modified to target protein-HLA complexes. Uveal melanoma cells overexpress gp100 protein and display gp100/HLA-A*02:01 complex on their surface. Tebentafusp-tebn binds this complex on the UM cells and bring them into contact with cytotoxic T-cells10,11 Binding of this ImmTAC molecule to T-cells result in T-cell activation and cytokine release, which lead to immune-mediated UM cell death.12
Clinical Trial Summary
A randomized, open-label, multi-center, phase III clinical trial assessed tebentafusp-tebn as first-line systemic treatment for metastatic UM.13 A total of 378 patients were randomly assigned in a 2:1 ratio to receive tebentafusp-tebn (n=252) or investigator choice therapy (n=126) consisting of pembrolizumab (n=106), ipilimumab (n=16), or dacarbazine (n=7). Tebentafusp-tebn was administered intravenously over 15-20 minutes at doses of 20 μg on day 1, 30 μg on day 8, and then 68 μg weekly while being monitored overnight due to risk of cytokine release syndrome after each of the first 3 doses. Key inclusion criteria were having HLA-A*02:01–positive tumors, no previous systemic or liver directed treatments, an Eastern Cooperative Oncology Group performance status score of 0 or 1, and at least one measurable lesion. Patients being treated with glucocorticoids for autoimmune disorder, displaying symptoms of central nervous system metastases, or receiving systemic immunosuppressive treatment were all excluded from this trial.13 The primary endpoint was overall survival analyzed in a time-to-event format. Secondary endpoints consisted of disease control categorized into complete response, partial response, or stable disease for more than 2 weeks as well as objective response (complete or partial response to therapy) and progression-free survival (measured on a time-to-event analysis). Safety was also evaluated.13
The primary endpoint of 1-year overall survival was 73% (95% confidence interval [CI], 66 to 79) in the tebentafusp-tebn group and 59% (95% CI, 48 to 67) in the control group (HR 0.51 [ 95% CI, 0.37 to 0.71]; P<0.001). The estimated median duration of overall survival for the tebentafusp-tebn arm was 21.7 months (95% CI, 18.6 to 28.6) compared to 16.0 months for the control group (95% CI, 9.7 to 18.4). Progression free survival at 6 months was higher with tebentafusp-tebn (31%) than in the control group (19%); HR=0.73; 95% CI, 0.58 to 0.94; P = 0.01. An objective response was seen in 9% of patients receiving tebentafusp-tebn and 5% of those in the control group. The median duration of response was 9.9 months in the tebentafusp-tebn group and 9.7 months in the control group. The percentage of patients who had disease control was 46% in the tebentafusp-tebn group (95% CI, 39 to 52) compared to 27% in the control group (95% CI, 20 to 36). Disease control was defined as a complete response, partial response or stable disease for ≥12 weeks.13
Authors from the trial reported that a majority of the treatment related adverse events stemmed from cytokine-related adverse events, such as pyrexia (76%), chills (47%), and hypotension (38%), and skin-related adverse events, such as rash (83%), pruritus (69%), and erythema (23%). Pre-medications to prevent adverse events were not allowed empirically and were employed if a patient experienced a side effect. A sub-analysis was performed to assess the overall survival benefit of developing a rash within 1 week of initiating tebentafusp-tebn. However, the sub-analysis did not support week 1 rash as an independent predictor of overall survival. Most patients experienced only grade 1 (12%) or 2 (76%) cytokine release syndrome and typically this presented within a few hours of their first dose. Only 1% of patients experienced grade 3 CRS and there were no reports of grade 4 or 5 CRS. Patients reported decreased symptoms of CRS as they progressed through the treatment cycles with majority of the patients reporting a decrease in adverse effects following the third week of dosing tebentafusp-tebn. Only 2% of patients in the tebentafusp-tebn group withdrew from the trial due to adverse effects. There were no treatment related deaths reported in this trial.13
Conclusion
The ImmTAC protein tebentafusp-tebn has the potential to emerge as the leading therapy for people diagnosed with HLA-A*02:01 positive metastatic UM. The Phase III trial demonstrated that treatment with this novel agent results in increased overall survival with a manageable adverse event profile compared to traditional cytotoxic chemotherapy. Future studies should address optimizing tebentafusp-tebn utilization and alternative treatment strategies to ensure improved patient outcomes.
REFERENCES
- Singh AD, Turell ME and Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118:1881–1885.
- Bishop KD and Olszewski AJ. Epidemiology and survival outcomes of ocular and mucosal melanomas: A population-based analysis. Int J Cancer. 2014 Jun 15;134(12):2961-71.
- Lane AM, Kim IK and Gragoudas ES. Survival rates in patients after treatment for metastasis from uveal melanoma. JAMA Ophthalmol. 2018 Sep 1;136(9):981-986.
- Smit KN, Jager MJ, Klein AD and Kilic E. Uveal melanoma: Towards a molecular understanding. Prog Retin Eye Res. 2020 Mar;75:100800.
- Carvajal RD, Schwartz GK, Tezel T, et al. Metastatic disease from uveal melanoma: treatment options and future prospects. Br J Ophthalmol. 2017;101:38–44.
- Eletr ZM and Wilkinson KD. An emerging model for BAP1’s role in regulating cell cycle progression. Cell Biochem Biophys. 2011 Jun;60(1-2):3-11.
- National Comprehensive Cancer Network. Melanoma: Uveal. (Version 2.2022), https://www.nccn.org/professionals/physician_gls/pdf/uveal.pdf. Accessed June 2, 2022.
- Damato BE, Desjardins L, Jager MJ, Kivelä T. (Eds.). (2011). Current Concepts in Uveal Melanoma (Vol. 49). S. Karger AG.
- Kimmtrak. Package Insert. Immunocore Limited; 2022.
- Damato BE, Dukes J, Goodall H and Carvajal RD. Tebentafusp: T Cell redirection for the treatment of metastatic uveal melanoma. Cancers. 2019; 11(7):971.
- Wagner SN, Wagner C, Schultewolter T and Goos M. Analysis of Pmel17/ gp100 expression in primary human tissue specimens: implications for melanoma immuno- and gene-therapy. Cancer Immunol Immunother. 1997 Jun;44(4):239-47.
- Oates J, Hassan NJ and Jacobsen BK. ImmTACs for targeted cancer therapy: Why, what, how, and which. Molecular Immunology. 2015;67(2): 67-74.
- Nathan P, Hassel JC, Rutkowski P, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med. 2021;385:1196- 206.