Novel Therapies in Sickle Cell Disease: Hype or Hope?
Madeleine Ochs, PharmD
Clinical Pharmacist Specialist, Inpatient Hematology
Michigan Medicine: University of Michigan
Ann Arbor, MI
Sickle cell disease (SCD) is an inherited red blood cell disorder, impacting roughly 100,000 people in the United States. SCD is caused by a single nucleotide substitution in the sixth codon of the beta-globin gene, which results in the substitution of valine for glutamic acid and the production of sickle hemoglobin (HbS). This substitution allows HbS to polymerize under deoxygenated conditions, distorting red blood cells into a sickled shape.1 Since the United States Food and Drug Administration (FDA) approval of hydroxyurea in 1998, the armamentarium for the treatment of SCD has been limited.
Although its mechanism is not fully understood, hydroxyurea is a ribonucleotide reductase inhibitor and increases synthesis of fetal hemoglobin. Fetal hemoglobin, which lacks beta-globin chains, inhibits sickling by interfering with the polymerization of HbS. The Multicenter Study of Hydroxyurea demonstrated that hydroxyurea significantly reduced the median annual rates of crises compared to placebo (median 2.5 vs 4.5 crises, p<0.001) in adults with ≥3 crises in the previous year.2 Long-term follow-up after 9 years found that hydroxyurea demonstrated a 40% reduction in mortality.3 Despite the mortality benefit, limitations with hydroxyurea include toxicity (myelosuppression, GI symptoms, infection, secondary malignancies) and poor adherence rates, partially due to patient perceptions and misconceptions.
Novel Therapies
Whereas there was only one approved treatment option over the last two decades, from 2017-2019 we saw a relative explosion of FDA approvals for the treatment of SCD, with 3 drugs approved: L-glutamine, crizanlizumab, and voxelotor (Table 1).
L-glutamine
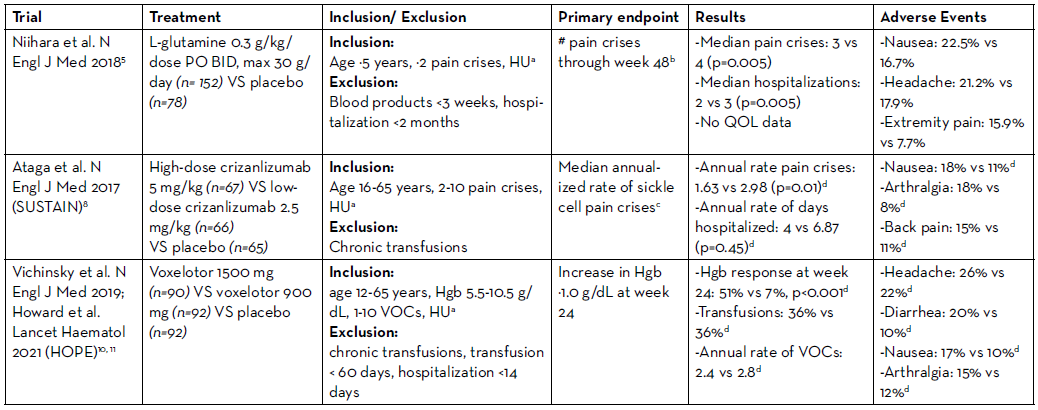
L-glutamine is a conditionally essential amino acid, which is required for the synthesis of NADPH, glutathione, and nitric oxide and becomes essential during times of oxidative stress, as in SCD.4 In 2017, L-glutamine received FDA approval based on the results of a phase III trial comparing L-glutamine 0.3 g/kg/dose by mouth (PO) twice daily (BID), max 30 g/day, to placebo in patients age ≥5 years who had ≥2 pain crises in the prior year.5 Patients were eligible if they were receiving hydroxyurea at a stable dose for ≥3 months, although hydroxyurea couldn’t be initiated or dose-escalated during the study. Roughly 2/3 of patients in both arms received concomitant hydroxyurea. Exclusion criteria included receipt of any blood products within three weeks prior to screening. The primary endpoint—the number of pain crises [defined as pain requiring parenteral narcotics or ketorolac in the ED/during hospitalization, acute chest syndrome (ACS), priapism, or splenic sequestration] through week 48—was significantly lower in the L-glutamine arm, with a median of 3 pain crises with L-glutamine vs 4 with placebo, p=0.005. L-glutamine also resulted in fewer hospitalizations (median 2 vs 3, respectively), but there was no difference in ED visits between arms. The discontinuation rate was higher in the L-glutamine arm at 36.2%, compared to 24.4% with placebo. The poor adherence rates with L-glutamine are unsurprising as it is supplied as a 5 g/packet oral powder, which is mixed with 240 ml of a cold/ room temperature beverage or 120-180 ml of food.6 Patients ≥65 kg receiving 15 g BID are therefore required to mix 3 packets two times daily. Because of the high dropout rate, the trial investigators imputed the pain crises as the mean of the patients in the same arm or the actual number of crises at the time of discontinuation carried forward for the duration of the 48 weeks, whichever was higher.
Due to the limitations of the statistical analyses, the FDA conducted their own sensitivity analyses and estimated that L-glutamine reduced the mean number of sickle cell crises from 0.4-0.9, compared to a difference of 1 in the published analysis. This study did not evaluate quality of life (QOL). Overall, L-glutamine was well tolerated; nausea, extremity pain, and back pain were more common in the L-glutamine arm. Despite the 0.3 g/kg twice daily dosing regimen studied in the phase 3 trial, the FDA approved dosing strategy is based on weight ranges (<30 kg: 5 g BID; 30-65 kg: 10 g BID; >65 kg: 15 g BID).
Crizanlizumab
Crizanlizumab is a humanized IgG2 kappa monoclonal antibody that binds P-selectin and prevents its interaction with P-selectin glycoprotein ligand 1 (PSGL-1), thereby decreasing the adhesion of sickled red cells.7 Crizanlizumab was FDA approved based on the results of the phase 2 SUSTAIN trial, which randomized 198 patients with SCD to low dose crizanlizumab (2.5 mg/kg), high dose crizanlizumab (5 mg/kg), or placebo IV every 2 weeks for 2 doses, followed by every 4 weeks for a total of 14 doses.8 Trial participants were 16-65 years old with 2-10 pain crises in the prior year. Again, patients receiving hydroxyurea were required to be at a stable dose for ≥3 months and hydroxyurea couldn’t be initiated or dose-escalated during the study. Patients receiving chronic transfusions were excluded. The primary endpoint, the median annualized rate of sickle cell pain crises (defined as acute episodes of pain resulting in treatment with PO/IV narcotics or IV NSAIDS at a medical facility, ACS, hepatic or splenic sequestration, or priapism) was significantly lower with high dose crizanlizumab at 1.63 vs 2.98 with placebo, p=0.01. There was no significant difference in the annual rate of days hospitalized, median rate of complicated crises including ACS, or QOL between arms. Again, due to the high discontinuation rate (35.8% high-dose, 31.8% low-dose, 36.9% placebo) the FDA performed sensitivity analyses.9 The decreased annual rate of vaso-occlusive crises (VOC) with high-dose crizanlizumab maintained significance when patients who discontinued the study early were excluded (1.18 vs 2.98 with placebo, p=0.005), however significance was lost when including only those who discontinued early.
Overall, crizanlizumab was well tolerated, and the most common adverse events in the high-dose group included nausea (18%), arthralgia (18%), headache (17%), extremity pain (17%), and back pain (15%). Serious adverse events (grade ≥3) in either crizanlizumab arm included pyrexia (n=2), influenza (n=3), and pneumonia (n=5). Infusion related reactions (occurring up to 24 hours post-infusion) occurred in 2 patients, and premedication with acetaminophen and diphenhydramine may reduce these reactions. A negative pregnancy test was required prior to initiation of crizanlizumab during the trial and incorporating pregnancy tests into order sets can help ensure negative pregnancy tests prior to treatment.
During the SUSTAIN trial, no antibodies against crizanlizumab were detected, however post-marketing studies required by the FDA are ongoing. Because crizanlizumab interferes with automated platelet counts resulting in platelet clumping when blood samples are collected using EDTA tubes, citrate tubes should be used to collect blood samples.7 Based on the results of the SUSTAIN trial, the FDA approved dose of crizanlizumab is 5 mg/kg.
Voxelotor
Voxelotor is a HbS polymerization inhibitor, which binds reversibly to hemoglobin and increases the affinity of hemoglobin for oxygen. Voxelotor was FDA approved in 2019 as a result of the phase 3 HOPE trial, which randomized patients to voxelotor 1500 mg or 900 mg PO once daily, or placebo.10 Eligible patients with SCD were age 12-65 years, with a hemoglobin of 5.5-10.5 g/dL, and had 1-10 VOCs in the prior 12 months. Similar to previous trials, hydroxyurea use was permitted if patients were on a stable dose for ≥3 months. Overall, 65% of patients were receiving hydroxyurea at baseline. Patients receiving chronic transfusions or those who received a transfusion in the previous 60 days were excluded. The primary endpoint was the percentage of patients with an increase in baseline hemoglobin by ≥1.0 g/dL at week 24, in contrast to the prior L-glutamine and crizanlizumab trials where pain crises were the primary endpoint. At week 24, voxelotor 1500 mg resulted in significantly more patients with a hemoglobin response compared to placebo (51% vs 7%, p<0.001; 33% with 900 mg voxelotor). With long-term follow-up, 89% of patients in the 1500 mg voxelotor arm, 72% with 900 mg, and 25% with placebo had an increase in hemoglobin by ≥1.0 g/dL at any time point by week 72.11
Despite the improvement in hemoglobin, there was no difference in the percentage of patients requiring RBC transfusions during the study, at 36% in all arms. Similarly, there was no significant difference in the annualized rate of VOCs (defined as pain requiring IV/PO opioids, ketorolac, or other analgesics or ACS) at 2.4 with voxelotor 1500 mg, 2.4 with 900 mg, and 2.8 with placebo. Based on a subgroup analysis, the authors claimed the incidence of VOCs was lowest among patients with the highest hemoglobin, ≥12 g/dL (n=10). Sickle cell anemia with crisis, ACS, priapism, and osteonecrosis were defined as sickle cell related events in contrast to the composite endpoint in the prior studies. There was no difference in the rate of sickle cell related adverse events among arms (78% 1500 mg, 75% 900 mg, 80% placebo), and grade 3 events were numerically higher in the 1500 mg arm (priapism: 6.5% 1500 mg, 2.4% 900 mg, 2.4% placebo; ACS 9.1% 1500 mg, 8.7% 900 mg, 6.6% placebo). In comparison to the previously discussed trials, the discontinuation rate was lower at 26% in the voxelotor 1500 mg arm, which may be attributed to the once daily dosing regimen. In the high dose voxelotor arm, the most common adverse events were headache (26%), diarrhea (20%), nausea (17%), and arthralgia (15%).
Voxelotor is a minor CYP3A4 substrate and weak CYP3A4 inhibitor and dose adjustments are recommended for use with concomitant CYP3A4 inducers or inhibitors.12 Based on the HOPE trial, the FDA approved dose of voxelotor is 1500 mg once daily. Despite PK studies with voxelotor 600 mg daily demonstrating mean AUC levels (concentrations adjusted for dose) were 90% higher with severe hepatic impairment, the package insert recommends dose adjustment to 1000 mg for severe hepatic impairment.13 Voxelotor may interfere with measurement of Hb subtypes and if needed, chromatography should be performed when patients are not receiving voxelotor.
Hype or Hope?
While it is encouraging there are now additional drug treatment options beyond hydroxyurea, the definition of acute pain crises, primary endpoints, and baseline characteristics differ across studies, which makes it difficult to determine the optimal place in treatment for these novel agents. The variable use of hydroxyurea and the inability to initiate or dose escalate hydroxyurea in the trials makes it challenging to determine what patients will benefit most from these novel therapies. The benefit of these agents in patients receiving chronic transfusions is unknown, as they were excluded from these trials. Furthermore, there is no long-term follow-up data available for these novel agents. There is no safety data in pregnancy as pregnant patients were excluded in all three trials. Few pediatric patients were included in these studies, with a minimum age of 5 years for L-glutamine, 12 years for voxelotor, and 16 years for crizanlizumab, however pediatric studies are ongoing.
Voxelotor recently received accelerated approval for use in children ages ≥4 years based on the Phase 2 HOPE-KIDS Study. Among the trials which examined QOL, there was no improvement demonstrated (not studied for L-glutamine). In addition to the limitations discussed above, these novel agents are associated with significant financial toxicity. Based on wholesale acquisition costs, a 30-day supply costs roughly $3,690 for L-glutamine, $9,642 for crizanlizumab, and $10,417 for voxelotor. This is especially relevant as these costs are likely prohibitive for patients who live in countries where the SCD burden is the highest.
As hydroxyurea has a demonstrated mortality benefit, these novel agents should not be used as a replacement for hydroxyurea, but rather in addition to hydroxyurea for patients with inadequate disease control. Instead, pharmacists can play a large role in helping increase adherence to hydroxyurea. L-glutamine and crizanlizumab could be considered for patients with ≥2 VOCs in the prior year and who are intolerant to or have a contraindication to hydroxyurea. However, the benefit of L-glutamine was impacted by the imputation method utilized to account for the high discontinuation rate. The role of voxelotor, if any, is limited to patients with symptomatic anemia but should be used with caution as it did not demonstrate any benefit on transfusion burden, VOCs, or QOL.
While there is a lot of hype with the novel therapies recently approved, I am hopeful for the number of investigational drugs for SCD in the pipeline. Allogeneic hematopoietic cell transplantation is the only curative option for SCD; however, its use is limited by the availability of suitable donors. Gene therapy is another potentially curative option and recent data demonstrated complete resolution of VOCs in patients who had ≥4 severe VOCs in the prior 2 years.14 Additional emerging therapies include incalcumab and GBT021601, which share mechanisms with crizanlizumab and voxelotor, respectively, but are administered at a reduced frequency/pill burden and agents with novel mechanisms such as compliment inhibition.
Table 1. Summary of Novel Agent Clinical Trials in Sickle Cell Disease
HU: hydroxyurea; Hgb: hemoglobin
aConcomitant hydroxyurea permitted if at a stable dose for ≥3 months; no initiation or dose-escalation allowed
bPain requiring parenteral narcotics or ketorolac in the ED/during hospitalization, ACS, priapism, or splenic sequestration
cAcute episodes of pain resulting in treatment with PO/IV narcotics or IV NSAIDS at a medical facility, ACS, hepatic or splenic sequestration, or priapism
dHigh-dose vs placebo
REFERENCES
- Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004 Oct 9-15 2004;364(9442):1343-60. doi:10.1016/S0140-6736(04)17192-4.
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. May 18 1995;332(20):1317-22. doi:10.1056/NEJM199505183322001.
- Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. Apr 02 2003;289(13):1645-51. doi:10.1001/jama.289.13.1645.
- Sadaf A, Quinn CT. L-glutamine for sickle cell disease: Knight or pawn? Exp Biol Med (Maywood). 01 2020;245(2):146-154. doi:10.1177/1535370219900637.
- Niihara Y, Miller ST, Kanter J, et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med. Jul 19 2018;379(3):226-235. doi:10.1056/ NEJMoa1715971.
- Endari. Package insert. Emmaus Medical I, 2017.
- Adakveo. Package insert. Novartis Pharmaceutical Corporation; 2019.
- Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 02 02 2017;376(5):429-439. doi:10.1056/NEJMoa1611770.
- Center for Drug Evaluation and Research. BLA Multi- Disciplinary Review and Evaluation, BLA 761128 ADAKVEO (Crizanlizumab-tmca). Accessed March 1, 2022 https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761128Orig1s000MultidisciplineR.pdf.
- Vichinsky E, Hoppe CC, Ataga KI, et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med. 08 08 2019;381(6):509-519. doi:10.1056/NEJMoa1903212.
- Howard J, Ataga KI, Brown RC, et al. Voxelotor in adolescents and adults with sickle cell disease (HOPE): long-term follow-up results of an international, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Haematol. May 2021;8(5):e323-e333. doi:10.1016/S2352- 3026(21)00059-4.
- Oxbryta. Package insert. Global Blood Therapeutics I, 2019.
- Preston RA, Marbury T, Balaratnam G, et al. Pharmacokinetics of Voxelotor in Patients With Renal and Hepatic Impairment. J Clin Pharmacol. 04 2021;61(4):493-505. doi:10.1002/jcph.1757.
- Kanter J, Walters MC, Krishnamurti L, et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N Engl J Med. 02 17 2022;386(7):617- 628. doi:10.1056/NEJMoa2117175.