HOPA Publications Committee
Bonnie Labdi, PharmD RPh, Chair
Ashley Glode, PharmD BCOP, Vice Chair
George Carro, RPh MS BCOP, Board Liaison
Jayde Bednarik, PharmD BCOP
Megan Bodge, PharmD
Megan Brafford, PharmD BCOP
Christine Gegeckas, RPh BCOP
Lindsay Hladnik, PharmD BCOP
Lisa Lohr, PharmD BSPharm BCOP BCPS
Katie Long, PharmD
David Reeves, PharmD BCOP
Lisa Savage, PharmD BCOP BCPS
Alexandra Shillingburg, PharmD
Candice Wenzell, PharmD BCOP
Board Update

Michael J. Vozniak, PharmD BCOP, HOPA President
HOPA had a busy and productive summer! In July, HOPA hosted its 4th Annual Industry Relations Council (IRC) Summit. Representatives from thirteen of the 15 IRC participants joined us in Chicago. HOPA provided an update on the organization, and board members led focused discussions on clinical pathways, oral chemotherapy, and patient advocacy. The summit was very informative and provided the board of directors with good insight to help move HOPA forward in the ever-evolving healthcare environment.
One of the most active and visible HOPA member benefits is the HOPA Listserv. The HOPA Listserv is a great forum for getting answers to practice questions and learning from others. However, the current Listserv has some limitations. The board reviewed and discussed alternative platforms that will provide a better member experience and more functionality and has decided to replace the Listserv with a product from Higher Logic. A few of the benefits of the Higher Logic product include
· a more comprehensive community experience
· resource libraries where members can post documents to share
· additional options for discussion delivery methods
· adaptive design, which allows users to fully interact on smart phones and tablets
· space for electronic advertising and association-related news.
Higher Logic’s functionality is broader than a Listserv’s. HOPA will assess how to best utilize the product for our members and work on the design and implementation in the months to come.
In other HOPA news, the Standards Committee has released the HOPA Investigational Drug Services Best Practice Standards document and will develop follow-up webinars to inform target audiences about these guidelines in the fall. In addition, the Standards Committee helped appoint a work group to review, comment on, and consider endorsing the U.S. Pharmacopeial (USP) Convention, General Chapter <800> Hazardous Drugs—Handling in Healthcare Settings Guideline. Ryan Forrey, Richard Cleveland, and Susan Spivey served on the work group and provided detailed comments that were sub- mitted to USP in July. The comments also have been posted on the HOPA website for member review.
The 2nd Annual HOPA Oncology Pharmacy Practice Management Program was held September 19–20 in Chicago. In addition to extending the program to 2 days, there was a preconference workshop, “Oncology Residency and Preceptor Program Development,” offered. We also are excited about HOPA’s upcoming 11th Annual Conference, which is being held March 25–28, 2015, in Austin, TX. Both programs offer great educational content, networking opportunities, and the chance to engage with pharmaceutical industry representatives in the exhibitor halls.
HOPA’s Health Policy Committee has been actively monitoring the Provider Status initiative and will soon reach out to the HOPA membership to solicit support. In addition, the Health Policy Committee traveled to Washington, DC, in September with our health policy consultants from the District Policy Group. The committee and our consultants met with members of Congress to seek their support for H.R. 4190 and recognize pharmacists as providers under Medicare Part B. Please stay tuned for opportunities to support H.R. 4190.
HOPA has created and appointed a new Recognition Committee. The committee will be led by Stephanie Sutphin, chair, and David DeRemer, vice chair. The committee is responsible for directing our HOPA membership awards program, which was the task of the former Nominations and Awards Committee. The committee will be charged with developing processes and procedures for a new HOPA Fellows Program. The goal is to recognize the first class of fellows at the 2016 Annual Conference.
As HOPA celebrates its 10th anniversary, it is an opportune time to reflect on the association’s progress over the years and to envision HOPA’s future. HOPA has enjoyed tremendous growth in our membership, engagement, and participation in both advocacy and the pharmacy profession in recent years. We continue to provide quality continuing education programs that have expanded in content and variety. HOPA’s growth and maturation is a result of the association fulfilling its goals and objectives outlined in the strategic plan revolving around the four goal areas: professional development, research, advocacy, and hematology/oncology pharmacy practice standards. HOPA’s strategic plan was created in 2010 and was revised in November 2012. As HOPA looks toward its future, the board of directors has decided to formally review and update the strategic plan in early 2015. This will allow the association to reflect on changes in our environment, assess our progress on our goals, and set or revise goals that work toward our envisioned future when all individuals affected by cancer have a hematology/oncology pharmacist as an integral member of their care team.
Brief Update on Newly Approved Agents for the Treatment of Chronic Lymphocytic Leukemia
Ashley Glode, PharmD BCOP
Clinical Oncology Pharmacy Specialist
PGY2 Oncology Residency Program Director
Adjunct Assistant Professor—Clinical Pharmacy and Outcomes Sciences Medical University of South Carolina/South Carolina College of Pharmacy Charleston, S
Megan V. Brafford, PharmD BCOP
Clinical Oncology Pharmacy Specialist Baptist Health Lexington
Lexington, KY
Chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) accounts for approximately 7% of newly diagnosed cases of non-Hodgkin’s lymphoma (NHL)1 and is the most common leukemia diagnosed in the Western world.2 CLL and SLL basically are the same disease and are treated similarly (unless otherwise indicated, however, this article will focus on SLL). With CLL, the disease burden primarily is in the bloodstream and bone marrow, and with SLL, the lymph nodes are involved.3 In the United States, 15,720 new diagnoses and 4,600 new deaths from CLL are predicted to occur in 2014.4 CLL is considered an indolent NHL with median age at diagnosis of 72 years.5 Signs and symptoms of this malignancy are vague and include weakness, weight loss, fever, night sweats, enlarged lymph nodes, and early satiety, but patients also may be asymptomatic when they are diagnosed.6
Diagnosis typically involves evaluation of the patient’s complete blood count (CBC) with differential, peripheral blood smear, immunophenotype of the circulating lymphocytes, and a thorough physical exam. Molecular cytogenetics are also performed to assess for specific gene mutations, such as deletion 11q or 17p, which are poor prognostic indicators. Unmutated IgVH (immunoglobulin heavy chain variable region) and high expression of Zap70 or CD38 also are poor prognostic factors. Bone-marrow biopsies are not required for diagnosis of CLL but may be completed in select patients. Excisional lymph node biopsies are required for the diagnosis of SLL.3,6,7
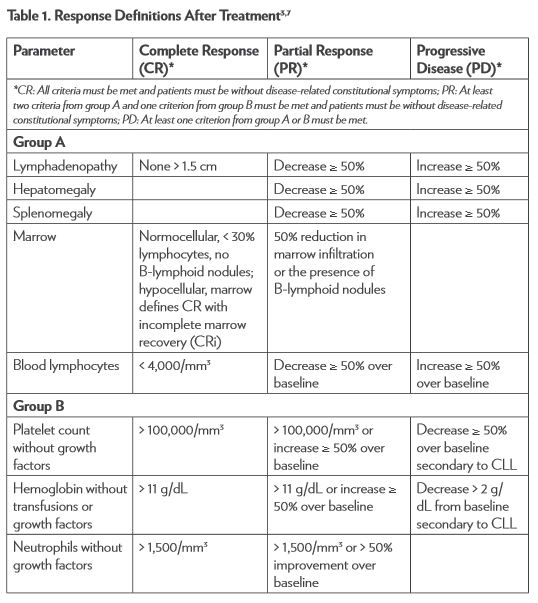
The National Cancer Institute–sponsored Working Group (NCI-WG) on CLL published revised guidelines for the diagnosis and management of this malignancy in 2008.7 To determine response to therapy (Table 1), assessment must include physical examination and evaluation of blood parameters. Response assessments should be conducted at least 2 months after treatment is completed. Stable disease (SD) is when patients do not have progressive disease (PD) but do not meet the criteria for complete response (CR) or partial response (PR). Relapse is described as evidence of disease progression after 6 or more months following an initial CR or PR. Refractory disease is expressed as failure to achieve a response or having disease progression within 6 months of the last treatment.3,7
Treatment Options
Treatment options for CLL have progressed during the past several decades, particularly in recent years. Several ongoing clinical trials are evaluating the efficacy of novel drug combination regimens and agents targeting unique pathways in B-cell malignancies. Treatment of this NHL subtype ranges from close observation with supportive-care measures to a variety of more intense therapeutic options. CLL is generally incurable, occurs in older patients, and progresses slowly. Therefore, it is often treated conservatively with careful consideration of the patient’s performance status and comorbidities.3,8
Patients who are asymptomatic may be observed but not treated until they become symptomatic, whereas patients with significant disease-related symptoms should be treated. Several pieces of clinical information should be considered if a patient is to be treated for CLL. Age, comorbidities, performance status, and presence of specific chromosomal abnormalities and gene mutations all should be evaluated when electing treatment regimens on an individual basis. Enrollment in a clinical trial should always be considered.3,6 Despite numerous available treatment options, some patients may be refractory to therapy, needing alternative treatment options, or require allogeneic hematopoietic stem cell transplantation (HSCT) for disease control. New agents have recently been added to the treatment armamentarium, which has improved patient options and prolonged survival.
Obinutuzumab (Gazyva)
Obinutuzumab is a humanized, glycoengineered IgG1 type 2 antibody targeted against CD20. Obinutuzumab has high-affinity binding for the type 2 epitope leading to 5- to 100-fold greater antibody-dependent cytotoxicity than rituximab.8,9 Obinutuzumab was approved by the U.S. Food and Drug Administration (FDA) in November 2013 for use in combination with chlorambucil for the treatment of patients with untreated CLL.10
Obinutuzumab was studied in a multinational trial (189 centers in 26 countries) that enrolled untreated patients with CD-20 positive CLL, Binet stage C, or symptomatic disease. Patients were also required to have a clinically meaningful burden of coexisting conditions, defined as a score higher than 6 on the Cumulative Illness Rating Scale (CIRS). Patients who did not have a high enough comorbidity score were also eligible if they had a creatinine clearance of 30–69 mL/min calculated with the Cockcroft-Gault formula. This open-label, three- group study randomized patients in a 1:2:2 manner: (a) chlorambucil alone, (b) obinutuzumab plus chlorambucil, and (c) rituximab plus chlorambucil, respectively; all regimens were given in six 28-day cycles. Chlorambucil was administered at 0.5 mg/ kg orally once on days 1 and 15. Obinutuzumab was administered at 1,000 mg intravenously on days 1, 8, and 15 during cycle 1, then only on day 1 during all subsequent cycles. The first infusion was divided over 2 days for cycle 1 after a protocol amendment to help decrease the rates of infusion reactions. Rituximab was administered at 375 mg/m2 intravenously on day 1 during cycle 1, then 500 mg/m2 during all subsequent cycles.9
After 118 patients had been assigned to the chlorambucil arm, this arm was closed early due to predefined stopping criteria. The protocol was revised to randomize patients in a 1:1 ratio into the remaining arms stratified by geographic region and stage. Patients in the single-agent chlorambucil arm were allowed to cross over to the obinutuzumab- plus-chlorambucil arm if they had PD during treatment or within 6 months of the end of treatment.9
A total of 781 patients were enrolled in the three study arms. Baseline characteristics among the three groups were well balanced, with a median age of 73 years, creatinine clearance of 62 mL/min, and a CIRS score of 8. The site investigator determined that progression- free survival (PFS) was the primary end point. There was significant improvement in PFS for the combination arms over the arm receiving chlorambucil alone—26.7 months for obinutuzumab plus chlorambucil versus 11.1 months for chlorambucil alone (hazard ratio [HR] = 0.18; 95% confidence interval [CI]: 0.13-0.24; p < .001) and 16.3 months for rituximab plus chlorambucil (HR = 0.44; 95% CI: 0.34-0.57; p < .001). Patients with deletion 17p were the only subgroup who did not experience this benefit in PFS. PFS was also significantly longer when obinutuzumab plus chlorambucil was compared with rituximab plus chlorambucil: 26.7 versus 15.2 months (HR = 0.39; 95% CI: 0.31-0.49; p <.001) . Additionally, obinutuzumab in combination with chlorambucil resulted in higher rates of overall, complete, and molecular responses.
At the time of publication, the most recent data for overall survival (OS) revealed a significant improvement for the obinutuzumab plus chlorambucil arm over the chlorambucil monotherapy arm; 9% versus 20% (HR for death= 0.41; 95% CI: 0.23-0.74; p = 0.002). There was no significant OS difference in the combination therapy arms.9
Adverse reactions occurred most commonly in the obinutuzumab plus chlorambucil arm, including neutropenia, anemia, thrombocytopenia, leukopenia, and infusion-related reactions. Infection grade 3–5 ranged from 11% to 14% and was not significantly different between groups, with the majority of infections being bacterial in nature. Twenty percent of patients experienced grade 3–4 infusion reactions with the first infusion of obinutuzumab, yet no grade 3–4 reactions occurred during subsequent cycles. Patients in the rituximab plus chlorambucil arm were the least likely of all groups to discontinue therapy early due to adverse events. The primary reason for discontinuation in the obinutuzumab plus chlorambucil group was infusion-related reactions, which decreased with the divided dosing on day 1 of cycle 1 (100 mg on day 1 and 900 mg on day 2).9
Obinutuzumab plus chlorambucil has been added to the National Comprehensive Cancer Network (NCCN) guidelines as a preferred treatment option for first-line therapy of CLL.3 The infusion-related adverse reactions are manageable with appropriate premedications of acetaminophen, antihistamine, and corticosteroid.9 Obinutuzumab is being evaluated in the relapsed/refractory setting as well as in various combinations in both the relapsed/refractory and untreated setting.11
Ibrutinib (Imbruvica)
Ibrutinib is an oral agent that inhibits Bruton’s tyrosine kinase (BTK). This enzyme target is essential for B-cell receptor signaling, proliferation, and survival.12,13 The FDA approved ibrutinib in February 2014 for the treatment of patients with CLL who have received at least one previous therapy.13 In July 2014, it was also approved for treatment of patients with deletion 17p CLL.14
Ibrutinib was evaluated in RESONATE, a phase 3, multicenter, open- label, randomized trial that enrolled patients with relapsed or refractory CLL or SLL. RESONATE compared ibrutinib, 420 mg orally once per day, with ofatumumab, 300 mg intravenously week 1 followed by 2,000 mg intravenously weekly for 7 weeks, then every 4 weeks for 16 weeks. From 67 sites in the United States, Australia, and seven European countries, 391 patients were stratified according to purine analog chemoimmunotherapy resistance and presence of 17p13.1 deletion. Due to positive results from the phase 2 trial with ibrutinib, the trial was revised to allow crossover of patients from ofatumumab to ibrutinib.13
The baseline characteristics of patients were well matched between the two groups. Patients in the ibrutinib group received a median of 8.6 (0.2–16.1) months of therapy, and patients in the ofatumumab group received 5.3 (0–7.4) months of therapy. The primary end point of PFS was significantly prolonged in the ibrutinib group: 9.4 months versus 8.1 months for the ofatumumab group (HR for progression or death = 0.22; 95% CI: 0.15–0.32; p < .001 by log-rank test). Ibrutinib’s impact on PFS was seen regardless of baseline clinical characteristics or molecular features. OS was also significantly prolonged in the ibrutinib arm (HR = 0.43; 95% CI: 0.24-0.79; p = .005). The improvement in OS was maintained in all subgroups according to the pretreatment and genetic abnormalities.13
Lymphocytosis occurred in 69% of patients in the ibrutinib arm and was not considered disease progression. This lymphocytosis is a result of the lymphocytes leaving the nodal compartments and resolves within 8 months in most patients.15 The most common nonhematologic adverse events occurring in at least 20% of patients were diarrhea, fatigue, pyrexia, and nausea in the ibrutinib arm and fatigue, infusion-related reactions, and cough in the ofatumumab arm. Grade 3 or higher adverse events occurring more often in the ibrutinib arm included diarrhea (4% versus 2%) and atrial fibrillation (3% versus 0%). Any grade bleeding-related adverse events were more common in the ibrutinib group (44% versus 12%). Additional adverse events more common in the ibrutinib arm included rash (8% versus 4%), pyrexia (24% versus 15%), infection (70% versus 54%), and blurred vision (10% versus 3%). Study treatment discontinuation due to adverse events occurred in 4% of patients in each arm.13
Ibrutinib is an effective therapy for patients with relapsed or refractory CLL/ SLL and patients with deletion 17p. It has been added to the most recent version of the NCCN guidelines as a category 1 recommendation for patients with relapsed or refractory disease.3 Ibrutinib is also being evaluated in untreated patients with CLL or SLL and in various combination therapies in the relapsed/refractory setting.11
Ofatumumab (Arzerra)
Ofatumumab is an IgG kappa human monoclonal antibody that binds to a distinct epitope composed of both small and large loops on the CD20 molecule. Ofatumumab has increased binding and more potent complement-dependent cytotoxicity than rituximab.16,17
Ofatumumab was initially approved by the FDA in October 2009 for the treatment of patients with CLL refractory to fludarabine and alemtuzumab on the basis of durable tumor reduction in a single-arm study. Ofatumumab was administered in eight weekly intravenous in- fusions followed by four monthly infusions with the first dose being 300 mg and doses 2 through 12 being 2,000 mg each. Patients experienced a 42% (99% CI: 26–60) investigator-determined objective response rate (ORR) and 6.5-month (95% CI: 5.8–8.3) median duration of response (DOR). All responses were partial.16,17
Ofatumumab received FDA approval for an additional indication in April 2014. It is now also approved for use in combination with chlorambucil for first-line treatment of CLL in patients for whom fludarabine-based therapy is considered inappropriate,18 based on results presented at the 2013 American Society of Hematology (ASH) Annual Meeting and Exposition.
A multicenter, randomized, open-label study was conducted in 447 patients randomized in a 1:1 manner comparing ofatumumab plus chlorambucil to chlorambucil alone. Patients were considered inappropriate for fludarabine-based therapy due to advanced age and/or comorbidities. Chlorambucil was administered at 10 mg/m2 orally on days 1 through 7 of each 28-day cycle, and ofatumumab was administered intravenously at 300 mg on day 1 and 1,000 mg on day 8 of cycle 1, followed by 1,000 mg on day 1 of subsequent cycles. Patients were treated for a minimum of three cycles, and treatment was continued until best response to a maximum of 12 cycles. Baseline demographics were well matched between treatment arms, with a median age of 69 years, 82% of patients aged 65 years or older, and/or having two or more comorbidities.19
The primary end point of PFS assessed by an independent review committee revealed a significantly longer PFS in the combination-therapy arm compared with the single-agent chlorambucil arm (22.4 versus 13.1 months; HR = 0.57, 95% CI: 0.45–0.73, p < .001). The secondary end point of overall response rate was also improved in the ofatumumab plus chlorambucil arm (82% versus 69%; OR 2.16; p = .001). CR rate was superior in the combination arm compared with the chlorambucil- alone arm: 12% versus 1%, respectively. At a median follow-up time of 29 months, the median OS was not reached for either arm; the trial concluded before survival time could be assessed. The median duration of treatment for both arms was six cycles, with 82% of patients receiving six or more cycles of ofatumumab plus chlorambucil.19
There were similar rates of grade 3 or higher adverse events occurring from the start of treatment through 60 days from the last dose (50% in ofatumumab plus chlorambucil versus 43% chlorambucil alone). The most common grade 3 or higher adverse event occurring in both groups was neutropenia (26% in the combination arm versus 14% in the single- agent arm) followed by infection (15% versus 14%, respectively). Ten percent of patients in the combination-therapy arm experienced grade 3 or higher infusion reactions despite premedication with acetaminophen, an antihistamine, and glucocorticoid; none were fatal.19
Ofatumumab is an important addition to the treatment options for patients with untreated CLL who are not candidates for fludarabine-based therapy. With CLL being diagnosed in older patients with comorbidities, this is an important advance in CLL therapy. Ofatumumab has not been added to the current version of the NCCN guidelines for this setting, but an update is in progress.3
Idelalisib (Zydelig)
Idelalisib is an oral, highly selective PI3K (PI3 kinase) inhibitor approved in July 2014. It is indicated for the treatment of relapsed CLL in combination with rituximab for patients for whom rituximab alone would be considered inappropriate treatment and as monotherapy for patients with relapsed SLL.20
Study 116 was a phase 3, randomized, double-blind, placebo-controlled trial conducted at 90 centers in the United States and Europe comparing idelalisib plus rituximab to placebo plus rituximab in patients with relapsed CLL. Patients were given idelalisib, 150 mg orally twice daily, or placebo with rituximab, 375 mg/m2 followed by 500 mg/m2 every 2 weeks for four doses and then every 4 weeks for three doses (a total of eight infusions). Patients were stratified by presence of 17p deletion or other TP53 mutations or the lack of IgHV mutation. Patients had to have been treated with a CD20 antibody or at least two previous cytotoxic regimens and be ineligible to receive cytotoxic therapy for any of the following reasons: severe neutropenia or thrombocytopenia from previous therapies, creatinine clearance less than 60 mL/min, or CIRS score higher than 6. Patients in the placebo group who experienced disease progression while enrolled in Study 116 were permitted to enroll in Study 117 to receive idelalisib. Patients with progression on idelalisib were allowed a dose increase to 300 mg orally twice daily.21
The groups were well matched with 110 patients randomized to each study arm. The median time on study was short because of early stopping parameters being met due to response. Patients received study treatment for 3.8 months in the idelalisib group and 2.9 months in the placebo group. Results were positive, with the idelalisib combination arm having a significantly improved primary end point of PFS (combination arm, not reached versus placebo arm, 5.5 months; HR = 0.15; p < .001), overall response (81% versus 13%; OR, 29.92; p < .001), and OS at 12 months (92% versus 80%; HR = 0.28; p = .02). Patients receiving idelalisib also experienced lymphocytosis, but this was lessened with the addition of rituximab. Lymphocytosis rates peaked at week 2 and resolved by week 12. Serious adverse-event rates were comparable between groups: 40% in the idelalisib plus rituximab group versus 35% in the placebo plus rituximab group. The most common adverse events in the idelalisib group were pyrexia, fatigue, nausea, chills, and diarrhea.21
Idelalisib’s accelerated approval for relapsed SLL is based on data from a single-arm, phase 2 study (101-09; DELTA) conducted at 41 U.S. and European sites in patients with relapsed indolent lymphoma refractory to rituximab and alkylating-agent containing chemotherapy. Idelalisib was administered at 150 mg orally twice daily until the disease progressed, unacceptable toxicities occurred, or the patient died. A total of 26 patients with SLL were included in this study, and they had an overall response rate of 58% (37%–77%), which was the primary end point. All 15 responses seen in patients with SLL were PRs with a median duration of response of 11.9 months (0–14.7 months). The median duration of treatment was 6.6 months (0.6–23.9 months), and the mean duration was 8.1 ± 5.7 months. The most common adverse events (≥ 20%) seen in all grades included diarrhea, fatigue, nausea, cough, and pyrexia.20,22
Idelalisib is an important addition to the available therapies for CLL/ SLL, having a distinctive mechanism of action. Idelalisib has yet to be added to the NCCN guidelines due to its recent approval and the guideline update currently in progress. It is included in several ongoing clinical trials in combination therapy and untreated patients.3,11,23
Future Directions
There are several new agents that have the potential to provide additional options for the management of CLL. The B-cell lymphoma 2 (Bcl-2) family of regulator proteins is highly involved in apoptosis and is a potential pathway to target in CLL because Bcl-2 is highly expressed in this disease. There are several small-molecule Bcl-2 inhibitors under investigation. The B-cell receptor (BCR) pathway is an additional potential target, as B cells rely on signaling mediated by BCR for maturation, proliferation, survival, and death. Some tyrosine kinases involved in this signaling include spleen tyrosine kinase (SYK), PI3K, and BTK. Inhibitors of these tyrosine kinases are already under investigation.3,11
Conclusion
CLL is a common subtype of NHL and is incurable with current treatment options outside of an allogeneic HSCT. Chemoimmunotherapy has improved OS for patients with CLL, but patients who experience relapsed or refractory disease continue to have poor outcomes. Because of the age of patients at diagnosis, it is important to consider several patient-specific factors and implement appropriate supportive-care measures when selecting a treatment option.3 Identifying treatment alternatives with improved side-effect profiles and patient tolerability is an important next step for management of this malignancy. The recently approved and in-development targeted therapies have the goal of filling this niche.
References
1. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood. 1997;89(11):3909-3918.
2.Altekruse SF, Kosary CL, Krapcho M, et al, eds. SEERCancer StatisticsReview,1975-2007.Bethesda, MD: National Cancer Institute. http://seer.cancer.gov/csr/1975_2007. Based on November 2009 SEER data submission, posted to the SEER website, 2010. Accessed August 11, 2014.
3.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Non-Hodgkin’s Lymphomas [Version 3.2014]. http://www.nccn.org/professionals/physician_ gls/pdf/nhl.pdf. Published July 18, 2014. Accessed August 11,2014.
4.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA CancerJClin.2014;64(1):9-29.
5.Eichhorst B, Dreyling M, Robak T, Montserrat E, Hallek M; ESMO Guidelines Working Group. Chronic lymphocytic leukemia: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. AnnOncol.2011;22(suppl 6):vi50-vi54.
6.Gribben JG. How I treat CLL up front. Blood. 2010;115(2):187-197.
7.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: areport from the International Workshop on Chronic LymphocyticLeukemia updating the National Cancer Institute-WorkingGroup 1996 guidelines. Blood.2008;111(12):5446-5456.
8.Robak T. GA-101, a third generation, humanized and glyco- engineered anti-CD20 mAb for the treatment of B-cell lymphoid malignancies. CurrOpinInvestigDrugs.2009;10(6);588-596.
9.Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chloarmbucil in patients with CLL and coexisting conditions. N Engl JMed.2014;371(12):1101-1110.
10.US Food and Drug Administration. Drugs: Grazyva (obinutuzumab). http://www.fda.gov/ Drugs/InformationOnDrugs/ApprovedDrugs/ucm373263.htm. Updated November 1, 2013. Accessed August 11, 2014.
11.National Institutes of Health, US Department of Health andHuman Services. ClinicalTrials.gov. http://clinicaltrials.gov/. July16, 2014.
12.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. NatRevCancer.2014(4);14:219-232.
13.Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. NEngl JMed.2014;371(3):213-223.
14.Imbruvica (ibrutinib) receives regular approval by US FDA in chronic lymphocytic leukemia (CLL) and CLL patients with del 17p: approval based on Phase 3 RESONATE data with statistically significant improvements in progression-free andoverall survival. Johnson & Johnson Web site. http://www.jnj.com/news/all/IMBRUVICA-ibrutinib-Receives-Regular-Approval-by-US-FDA-in-Chronic-Lymphocytic-Leukemia-CLL-and-CLL-patients-with-del-17p. Published July 28, 2014. Accessed August11, 2014.
15.Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate suboptimal response to therapy. Blood.2014;123(12):1810-1817.
16.Lemery SJ, Zhang J, Rothmann MD, et al. US Food andDrug Administration approval: ofatumumab for the treatment of patients with chronic lymphocytic leukemia refractory to fludarabine and alemtuzumab. ClinCancer Res.2010;16(17):4331-4338.
17.Wierda WG, Kipps TJ, Mayer J, et al. Ofatumumab as single- agent CD20 immunotherpay in fludarabine-refractory chronic lymphocytic leukemia.JClinOncol.2010;28(10):1749-1755.
18.US Food and Drug Administration. Ofatumumab. http://www.fda.gov/ Drugs/ InformationOnDrugs/ApprovedDrugs/ucm393823.htm. Updated April 17, 2014. Accessed August 11,2014.
19.Hillmen P, Robak T, Janssens A, et al. Ofatumumab + chlorambucil versus chlorambucil alone in patients with untreated chronic lymphocytic leukemia (CLL): results of the phase III study complement 1 (OMB110911). Paper presented at: American Society of Hematology 55th Annual Meeting and Exhibition; December 7-10, 2013; New Orleans, LA. Abstract 528. https:// ash.confex.com/ash/2013/webprogram/Paper58498.html. Accessed August 11, 2014.
20.US Food and Drug Administration approves Gilead’s Zydelig® (idelalisib) for relapsed chronic lymphocytic leukemia, follicular lymphoma and small lymphocytic lymphoma [news release]. Foster City, CA: Gilead Sciences, Inc. July 23, 2014. http://investors.gilead.com/phoenix.zhtml?c=69964&p=irol-newsArtic le&ID=1950339&highlight=\#sthash.SYMspXkc.dpuf. Accessed August 11, 2014.
21.Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. NEngl JMed.2014;370(11):997-1007.
22.Gopal AK, Kahl BS, de Vos S, et al. PI3K inhibition by idelalisib in patients with relapsed indolent lymphoma. NEngl JMed.2014;370(11):1008-1018.
23. US FDA accepts new drug application for Gilead’s idelalisib for the treatment of refractory indolent non-Hodgkin’s lymphoma [news release]. Foster City, CA: Gilead Sciences, Inc. January 13, 2014. http://www.gilead.com/news/press-releases/2014/1/us-fda-accepts-new-drug-application-for-gileads-idelalisib-for-the-treatment-of-refractory-indolent-nonhodgkins-lymphoma. Accessed August 11, 2014.
HOPA Publishes Investigational Drug Service Best Practice Standards
We are pleased to announce the HOPA Standards Committee has completed the HOPA Investigational Drug Service Best Practice Standards, the first of its kind that provides the best practice standards and guidance for pharmacists and institutions that conduct clinical trials. The HOPA Investigational Drug Service Best Practice Standards emphasizes the critical role of the pharmacist in the investigational drug service from protocol concept to close-out. This document provides a foundation for pharmacy to be involved very early in protocol development and review, to ensure that the trial meets institutional medication guidelines, is executed efficiently, and adheres to all regulations and standards. In addition, the guidelines provide an outline for the role of the pharmacy technician which is unique to this document. These guidelines address various best practices for pharmacy operations and provide ancillary information about the different mechanisms for obtaining investigational drugs for a single patient, and provide a concise summary of and reference source for the procedures for obtaining investigational drugs on a “compassionate” basis. This best practice guideline should be used in conjunction with other applicable state and federal guidelines.
HOPA wishes to thank the following members for their valuable contributions:
Editor
Barry Goldspiel, PharmD BCPS BCOP
Lead Authors
Sapna Amin, PharmD BCOP
Joyce Lee, PharmD BCOP BCPS
Authors
Jorge Avila, PharmD BCOP
Matthew Boron, RPh
Stefanie Conley, PharmD RPh
Robert Enos, RPh MBA
Kathy Galus, PharmD BCOP
Theresa Mays, PharmD BS BCOP FASHP
Gopal Patil, PhD MBA RPh
Jennifer Thompson, PharmD BCOP CCRP
Peer Reviewers
Linda Bressler, PharmD BS
Michael Kane, RPh BCOP
Jennifer LaFollette, PharmD BCOP
Susan Rogers, RPh
You can access the HOPA Investigational Drug Service Best Practice Standards on HOPA’s website.
2014 ASCO Annual Meeting Review
Michael Newton, PharmD BCOP
Clinical Associate Professor
West Virginia University School of Pharmacy Clinical Specialist, Ambulatory Oncology West Virginia University Healthcare
Morgantown, WV
The American Society of Clinical Oncology (ASCO) held its 50th Annual Meeting in Chicago, IL, May 31–June 3, 2014. The official theme of this year’s meeting was “Science and Society” and focused on opportunities for the community of clinicians and researchers to lead society in the quest for knowledge and insight as it pertains to cancer. The meeting certainly delivered on its theme, presenting a diverse array of trial results that will affect practice and guide future research. The following is a summary of a few of the important findings presented.
LBA1: Adjuvant Exemestane with Ovarian Suppression in Premenopausal Breast Cancer Patients
The combined results of two phase 3 trials, Tamoxifen and Exemeastane Trial (TEXT) and the Suppression of Ovarian Function Trial (SOFT), were presented during the plenary session. The trials compared 5 years of adjuvant exemestane plus ovarian suppression with tamoxifen plus estrogen suppression in 4,690 premenopausal women. The method of ovarian suppression was based on physician and patient preference and included triptorelin, oophorectomy, or ovarian irradiation. Disease-free survival at 5 years was superior in the exemestane group (91.1% versus 87.3%; hazard ratio [HR] for disease recurrence, second invasive cancer, or death = 0.72; 95% confidence interval [CI]: 0.60–0.85; p < .001).
LBA4: Disappointing Final Results of ALTTO Trial
This trial examined the benefit of adding lapatinib to trastuzumab either concurrently or sequentially, in the adjuvant treatment of HER2-positive breast cancer. Unfortunately, lapatinib did not increase disease-free survival compared with trastuzumab alone. These results were disappointing and unexpected given the significantly increased pathologic complete response rates reported from the addition of lapatinib to trastuzumab in the neoadjuvant setting (reported previously in the NeoALTTO trial, available in Lancet 2012;379:633-640).
LBA505: Prevention of Early Menopause Study (POEMS)
In this trial, women younger than 50 years with stage I, II, or IIIa ER/ PR-negative breast cancer were randomized to receive a cyclophosphamide-based adjuvant chemotherapy regimen with or without goserelin at a dose of 3.6 mg monthly, starting 1 week before chemotherapy. The primary endpoint was rate of premature ovarian failure (POF). Rate of POF was 22% in the standard arm and 8% in the goserelin arm (OR = 0.30, 95% CI: 0.10–0.87; p = .03). A secondary endpoint in this trial was rate of pregnancy. Roughly the same proportion of women in each arm reported that they attempted to conceive after treatment, and more women in the goserelin arm were able to become pregnant (21% versus 11%; OR = 2.45; p = .03).
LBA 9500: Zoledronic Acid Every 4 Weeks Versus Every 12 Weeks
In this prospective, randomized, double-blind trial, women with bone metastases from breast cancer who previously received approximately 1 year of monthly intravenous (IV) bisphosphonate (either zoledronic acid or pamidronate) were randomized to receive either zoledronic acid 4 mg intravenously every 4 weeks or every 12 weeks for 1 year. There were no differences between the arms in rates of skeletal-related events. Treatment-related adverse events were similar in both arms; however, there were two cases of osteonecrosis of the jaw in the every-4-week arm and none in the every-12-week arm.
Abstracts 8002 and 8003: Important Data for Patients with ALK Translocation Positive NSCLC
In abstract 8002, the PROFILE 1014 trial confirmed that in first-line treatment, crizotinib is superior to doublet chemotherapy in patients with advanced anaplastic lymphoma kinase (ALK)-positive, nonsquamous non-small cell lung cancer (NSCLC). The study met its primary objective of prolongation of progression-free survival (PFS), demonstrating superiority of crizotinib over chemotherapy (median PFS 10.9 versus 7.0 mo; HR = 0.454; 95% CI: 0.346−0.596; p < .0001). The overall response rate was also significantly higher with crizotinib (74% versus 45%; p < .0001). Overall survival data are not yet mature, however, overall survival is a secondary endpoint, and the trial has a crossover design.
Abstract 8003 reported results from the expansion phase of the ASCEND-1 trial of ceritinib (formerly LDK378) in ALK-positive lung cancer patients (phase 1 results available in NEJM 2014;370:1189-1197). In the 180 patients evaluated, overall response rate was 60%. Of the 121 patients who had previously received crizotinib, the response rate was 55.4%, with a median duration of response of 7.4 months. Ceritinib gained U.S. Food and Drug Administration (FDA) approval in April 2014 for treatment of ALK-positive lung cancer patients previously treated with crizotinib.
Abstract 8005: Erlotinib Plus Bevacizumab Versus Erlotinib Alone as First-Line Treatment for Advanced EGFR Mutation-Positive Nonsquamous NSCLC
This open-label trial randomized epidermal growth factor receptor (EGFR)–positive advanced or metastatic NSCLC patients to receive either erlotinib 150 mg by mouth daily or the same dose of erlotinib combined with bevacizumab 15 mg/ kg intravenously every 3 weeks. Progression-free survival was superior in the combination arm (16.0 months versus 9.7 months; HR, 0.54; 95% CI: 0.36–0.79; log-rank p = .0015). Response rates were similar, and adverse events were considered manageable in the combination arm.
LBA2: Impact on Overall Survival with Chemohormonal Therapy Versus Hormonal Therapy for Hormone-Sensitive Newly Metastatic Prostate Cancer: An ECOG-Led Phase 3, Randomized Trial
In this potentially practice-changing trial, 790 men with metastatic hormone-sensitive prostate cancer who received androgen-deprivation therapy (ADT) were randomized to ADT alone or ADT plus docetaxel 75 mg/m2 every 3 weeks for six cycles. Patients receiving docetaxel were required to start chemotherapy within 4 months of initiation of ADT. The primary endpoint of overall survival demonstrated superiority of combination chemohormonal therapy compared with ADT alone (median overall survival was 52.7 months versus 42.3 months; p = .0006). Of note, patients who progressed on the ADT-alone arm were eventually given docetaxel as per the current standard of care. Survival benefit was most profound in men with high-volume disease, with an increase in survival of 17 months. Data are currently insufficient to determine whether men with low-volume disease would benefit from this strategy.
LBA3: CALGB/SWOG 80405 Phase 3 Trial of FolFIRI or FolFOX with Bevacizumab or Cetuximab for Patients with KRAS Wild-Type Untreated Metastatic Colorectal Cancer
Results from the CALGB/ SWOG 80405 trial (LBA3) found that for first-line treatment of metastatic colorectal cancer in patients who are KRAS wild-type, overall survival for cetuximab plus chemotherapy is equivalent to bevacizumab plus chemotherapy (approximately 29 months in both arms). It should be noted that the selection of FolFOX or FolFIRI as the first-line chemotherapy backbone was at the discretion of the treating physician and that there was a strong preference for FolFOX (73.4%). This is particularly important because previous studies have suggested that the cetuximab plus FolFOX combination may be inferior to FolFOX alone in the first-line setting.
As usual, the ASCO Annual Meeting presented clinicians with an enormous amount of data to digest. While not discussed in this review, excitement about immunotherapy continued to grow at ASCO 2014. We will likely see the first of the PD-1 inhibitors approved in late 2014, and I encourage readers to review abstracts related to these agents in melanoma and in other disease states. Also of note, value in cancer care was a recurrent theme at the meeting, which is not surprising given ASCO’s recent launch of the strategic Value in Cancer Care Initiative.
Recalls and Safety Alerts from the FDA
Exemestane (Aromasin)
The “Warnings and Precautions” section for exemestane now recommends that women with osteoporosis or those at risk of osteoporosis undergo a formal assessment of their bone mineral density by bone densitometry when initiating therapy. Patients should be monitored for bone mineral density loss and treated as indicated. Additionally, postmarketing reports of paresthesia and acute generalized exanthematous pustulosis have been added to the prescribing information.
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ucm250607.htm
Crizotinib (Xalkori)
The “Clinical Pharmacology Drug Interactions” section of the package insert has been revised to include information regarding the potential for crizotinib to inhibit uridine diphosphate glucuronosyl-transferase (UGT) enzymes, as well as other hepatic and renal transporters. Additionally, updated safety margins for pregnant and pediatric patient populations have been included within the “Use in Specific Populations” section of the package insert.
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ucm295722.htm
Docetaxel Intoxication
The U.S. Food and Drug Administration (FDA) has issued a safety announcement to warn that docetaxel contains ethanol and may cause patients to feel intoxicated or drunk during and after treatment. Product label revisions are being made to warn about the risk. Healthcare professionals (HCPs) should take this into consideration when prescribing and administering docetaxel, especially in patients in whom alcohol intake should be avoided or minimized, and when used in combination with certain drugs. Patients should be monitored for signs of alcohol intoxication during and after treatment. Alcohol content may vary between formulations. In patients who experience this adverse reaction, HCPs may consider using a docetaxel formulation with the lowest alcohol content and slowing the infusion rate during administration. Patients should be notified about the alcohol content in docetaxel and the potential for this to affect the central nervous system. Patients should be advised to avoid driving, operating machinery, and performing activities that are dangerous or require skill for 1 to 2 hours after the infusion.
http://www.fda.gov/ Drugs/ DrugSafety/ucm401752.htm
Gemcitabine (Gemzar)
The “Warnings and Precautions,” “Adverse Reactions,” and “Dose Modifications” sections of gemcitabine’s prescribing information have been updated to include information on the risk for posterior reversible encephalopathy syndrome (PRES). PRES has been reported with single-agent gemcitabine, as well as in combination with other chemotherapy agents. If PRES develops during therapy with gemcitabine, it is recommended to permanently discontinue the agent.
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ucm356107.htm
Denosumab (Xgeva) Hypocalcemia with Renal Dysfunction
The prescribing information for denosumab (Xgeva) has been revised to update information on the risk for hypocalcemia in the “Warnings and Precautions” and “Use in Specific Populations” sections. There is an increased risk for the development of hypocalcemia in patients with renal dysfunction, most commonly in patients with severe impairment—defined as those with a creatinine clearance less than 30 mL/min and/or those on dialysis—and with inadequate or no calcium supplementation. Calcium levels should be monitored and supplemented with calcium and vitamin D as needed.
http://www.fda.gov/safety/medwatch/safetyinformation/ucm343116. htm
Denosumab (Prolia) Musculoskeletal Pain
The “Warnings and Precautions” section of the prescribinginformation for denosumab (Prolia) has been revised to include postmarketing reports of severe and possibly incapacitating bone, joint, and muscle pain. Severe symptoms may warrant therapy discontinuation. http://www.fda.gov/safety/medwatch/safetyinformation/safety-relateddruglabelingchanges/ucm307218.htm
Eculizumab (Soliris) Recall
Alexion Pharmaceuticals, Inc., has issued a voluntary recall of certain lots of eculizumab distributed in the United States. This was due to the use of a process component during vial filling that resulted in the presence of visible particles. The company has identified the problem and is implementing a process change. There have been no safety risks identified in patients who have received eculizumab. There are no anticipated interruptions in patient supply.
http://www.fda.gov/safety/recalls/ucm399527.htm
Sunitinib (Sutent) Proteinuria and Dermatologic Toxicities
Proteinuria and dermatologic toxicities, including Stevens-Johnson syndrome, erythema multiforme, toxic epidermal necrolysis,and necrotizing fasciitis, have been added to the “Warnings and Precautions and Medication Guide” of sunitinib’s package insert.
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ucm224050.htm
Docetaxel Respiratory Adverse Reactions
There have been additional respiratory adverse reactions reported in postmarketing surveillance of docetaxel. The package insert has been updated and now includes the following statement: “Dyspnea, acute pulmonary edema, acute respiratory distress syndrome/pneumonitis, interstitial lung disease, interstitial pneumonia, respiratory failure, and pulmonary fibrosis have rarely been reported and may be associated with fatal outcome. Rare cases of radiation pneumonitis have been reported in patients receiving concomitant radiotherapy.”
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ ucm396551.htm
Temozolomide (Temodar) Hepatotoxicity
The “Warnings and Precautions” section for temozolomide’s package insert has been updated to include the risk of fatal and severe hepatotoxicity reported in patients receiving this agent. Recommended monitoring includes liver function tests at baseline, midway through the first cycle, prior to each subsequent cycle, and approximately 2 to 4 weeks after the last dose of temozolomide. A case control study is being conducted to determine the correlation between temozolomide and severe acute liver injury.
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ucm211909.htm
Thalidomide Venous and Arterial Thromboembolism Update
Updates have been made to the “Warnings and Precautions—Venous and Arterial Thromboembolism” section of the package insert for thalidomide. The update includes the following information: “Ischemic heart disease (11.1%), including myocardial infarction (1.3%) and stroke (cerebrovascular accident, 2.6%) have also occurred in patients with previously untreated mutliple myeloma treated with Thalomid and dexamethasone compared to placebo and dexamethasone (4.7%,1.7%, and 0.9%, respectively) in one clinical trial.”
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ucm402899.htm
Topotecan (Hycamtin) Renal Impairment
Revisions to the dosing recommendations in renal impairment for oral topotecan have been made in the package insert.
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ucm279915.htm
Pazopanib (Votrient) Drug Interactions
The solubility of pazopanib is pH dependent. Concomitant administration of pazopanib with drugs that raise gastric pH should be avoided. A drug interaction trial demonstrated a decrease in the exposure of pazopanib by approximately 40% (AUC and Cmax) when administered with esomeprazole. If therapy with these agents is necessary, short-acting antacids instead of PPIs and H2 receptor antagonists should be used whenever possible. The administration of antacids and pazopanib should be separated by several hours.
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ucm303649.htm
Obinutuzumab (Gazyva) Thrombocytopenia and Hemorrhagic Events
Updated safety data include reports of fatal hemorrhagic events during cycle 1 in patients treated with obinutuzumab. It is recommended to monitor all patients frequently, especially during the first cycle, for thrombocytopenia and hemorrhagic events. Dose delays of obinutuzumab and chlorambucil or dose reductions of chlorambucil can be considered in patients with grade 3 or 4 thrombocytopenia. Consideration should be made to withholding concomitant agents that increase bleeding risk, especially during the first cycle of therapy.
http://www.fda.gov/ Safety/MedWatch/ SafetyInformation/ucm404996.htm
ISMP Medication Safety Alerts (March–June)
May8,2014 (Volume19,Issue9)
The Institute for Safe Medication Practices (ISMP) has asked the FDA and Teva to investigate commas that have replaced decimals on tbo-filgrastim (Granix) syringes. The outer carton and peel-away prefilled syringe wrappers list the syringe volume using a decimal point (e.g., 300 mcg per 0.5 mL). However, the barrel of the syringe uses commas for volume increments (e.g., 0,5 mL). The use of a comma rather than a decimal has caused problems in the past. Toprevent inadvertent errors from occurring, ISMP has requested further investigation.
May22,2014 (Volume19,Issue10)
The ISMP reported a dosing error that occurred with nilotinib (Tasigna). A patient was instructed to take once-daily dosing because of previous intolerance to twice-daily dosing. The pharmacy filledthe prescription with instructions to take once daily. However, the preprinted dosing instructions on the manufacturer’s blister packaging provide instructions to take the medication every morning and evening. This led to confusion; the patient took nilotinb twice daily instead of once daily. The ISMP has notified the FDA and Novartis about the incident.
Resident’s Cubicle: Research Projects
Megan Bodge, PharmD
Stem Cell Transplant Clinical Specialist
VA Tennessee Valley Healthcare System
Nashville, TN
The residency year starts each July with a new crop of bright-eyed oncology pharmacy residents and a flurry of activity to prepare for the coming year. Residents often are expected to complete multiple projects throughout their 1-year residency—from medication usage evaluations to administrative projects to their main research project. They usually are expected to hit the ground running with early project selection and institutional review board (IRB) submission, all while getting acclimated to new practitioners, computer systems, and institutional practices. This edition of The Resident’s Cubicle will focus on tips to help residents with their post-graduate year 2 (PGY-2) research projects.
Residents entering their PGY-2 should be comfortable completing various projects given experiences from their first year of residency; however, they should be ready for the increased expectations and demands of PGY-2. Projects completed in the first year may not have been oncology focused and may have been the resident’s first experience with completing a major research project. With the transition to PGY-2, residents should be prepared to complete a high-quality project, which may require broadening their oncology knowledge base. Preceptors may also expect that the project will be completed with more independence and at a higher level than was expected during the previous year. Residents should be prepared to undertake a project that holds potential benefits for their own learning, their institution, and, ideally, oncology pharmacy practice. Although this may seem overwhelming at the beginning of the year, breaking the project down into smaller steps that can be accomplished throughout the year can make it more manageable.
Project Selection
Most residents will be presented with a list of possible project ideas at the beginning of their PGY-2—a result of preceptor brainstorming during the prior year. The number and type of team members (physicians, pathologists, nurses, etc.) involved with the project will vary based on the complexity of the project and subject matter. It is important that all of the key practitioners are involved; having a large research team can be helpful when brainstorming ideas and delegating project tasks. However, a large team also can be challenging because it is difficult to please everyone when ideas differ among team members. An additional challenge for incoming residents at a different institution than in their first year is getting a good feel for the preceptors and practitioners with whom they will be working on each project. Prior residency alumni are a great resource and often are willing to give candid advice about strengths and weaknesses of specific projects or preceptors. It is important to ensure that the resident selects a preceptor with whom they feel comfortable, because the project will require frequent interactions with other project team members and open communication at all times. Ultimately, it is important for all parties to remember that this project is the resident’s, and he or she should have the final say in project and research team selection.
Many oncology residents enter their second year of training with a specific area of focus for their residency year and, potentially, their career. However, with the uncertainty of the job market from year to year, it is important that residents’ projects are diverse and that they exit the year as well-rounded oncology pharmacy clinicians. If the resident decides to complete a solid tumor medication usage evaluation, he or she may want to consider a research topic that is in another area, such as hematology or stem cell transplant. Ultimately, it is important for the resident to be passionate about the topic he or she chooses. The major research project will require countless hours to complete, and the project quality will likely correlate with the interest level the resident has. In addition, a resident will be most proud of a project that holds meaning for him or her.
Last, when considering project ideas, it is important for the resident to consider the feasibility of project completion within a 1-year time frame. Feasibility is often incorporated into project idea review by residency preceptors prior to the PGY-2 resident’s arrival; however, the true feasibility of an individual project will vary based on the caliber of the resident and his or her time management skills. Residents often are ambitious and want to complete meaningful, large-scale projects. Ambition in residents is highly desirable; however, it is very undesirable if the project cannot be completed as planned. Meetings with the entire project team at the beginning of the year can help establish timelines and outline project expectations to ensure the project is completed as planned.
Project Timeline
Establishing a timeline for project milestones—such as IRB submission, completion of data collection, meeting with statisticians, abstract submission to a national meeting, and manuscript preparation—can be extremely helpful for staying on track throughout the year. During the first year of residency, residents often focus on presentation of their project at a regional residency conference. One difference that PGY-2 oncology residents may face is the shortened time frame for project completion if the resident is expected to present his or her project results at the HOPA Annual Meeting in March. Residents should meet with his or her program director and research team early in the year to decide where the project will be presented, and the resident should adjust his or her timeline accordingly.
The research team also should decide early on whether manuscript submission to a peer-reviewed journal is the ultimate goal, and, if so, the target journal should be determined. There may be multiple journals to consider based on what the research project aims to accomplish. Members of the team may already have a target journal in mind; however, if that is not the case, the resident should research journals to determine the most appropriate one based on journal scope and impact factor, design of the research project, and project findings. Selecting the target journal early will be helpful to guide formatting when preparing the manuscript. Even if the project does not end up showing a significant change or difference, it is still important to consider submitting the manuscript to inform other practitioners and institutions of the findings.
Residents should schedule regular meetings with other project team members throughout the year to ensure adherence to the project timeline. It also may be advisable to keep minutes from each meeting and e-mail them to team members to make sure everyone is on the same page. It is important that IRB submission be completed early in the year because unexpected delays are a common obstacle. Most importantly, any delays the resident encounters during the year should be quickly communicated to the rest of the research team. Residency project mentors are selected to teach research skills and guide the res- ident through the project. Although more independence may be expected from a PGY-2 resident, mentors can likely help get the project back on track as long as there is open communication at all times and willingness from both parties to stay actively involved.
Take-Home Points
The PGY-2 pharmacy research project should be meaningful and contribute to the advancement of oncology pharmacy practice. When the project has been completed, it is important that these data are presented to the institution’s hospital staff. The goal behind completing the project is usually to improve or change a process at the hospital, and disseminating the findings will hopefully contribute to improving patient care. If a change is implemented as a result of the project, that information could be included in the manuscript so that other institutions can determine whether a similar change would be beneficial. Ultimately, the project may have the potential to make an impact both locally and nationally.
Residents should not lose track of the fact that this project is also a learning experience. Although project outcomes are certainly important, the research skills that can be learned from completing the project, no matter the topic, are at least as valuable. Residents will soon find themselves in the role of preceptor and project mentor to other residents and students. It is imperative that residents take full advantage of the top- notch physicians and experienced pharmacist preceptors with whom they have the opportunity to work and absorb all of the wisdom and knowledge that can be learned throughout the year.
Although it may be overwhelming to consider at the beginning of the year the entirety of the project that needs to be completed, focusing on smaller aspects of the research project timeline can help to restore sanity. The demands of PGY-2 certainly keep residents busy, and the year will fly by. Before residents realize it, they will be presenting the results of their long hours of research to their colleagues and wondering where the year went. I urge current residents to take advantage of meetings attended throughout the year to network with other current and future hematology/oncology pharmacists. Research projects require a lot of hard work, and maybe a sleepless night or two, but aspects of the project can certainly be fun, too!