HOPA Publications Committee
Christan M. Thomas, PharmD, BCOP, Editor
Lisa Cordes, PharmD, BCOP, BCACP, Associate Editor
Renee McAlister, PharmD BCOP, Associate Editor
Lydia Benitez, PharmD, BCOP
Alexandra Della Pia, PharmD, MBA, BCOP
Jeff Engle, PharmD, MS
Karen M. Fancher, PharmD, BCOP
Chung-Shien Lee, PharmD, BCOP, BCPS
Robert Steven Mancini, PharmD, BCOP, FHOPA
Bernard L. Marini, PharmD, BCOP
Alan L. Myers, PharmD, PhD
Gregory T. Sneed, PharmD
Diana Tamer, PharmD, BCOP
Kristin Held Wheatley, PharmD, BCOP
HOPA News Staff
David DeRemer, PharmD, BCOP, FCCP, FHOPA, Board Liaison
Michelle Sieg, Communications Director
Joan Dadian, Marketing Manager
View PDF of HOPA News, Vol. 19, no. 1
Board Update: Moving HOPA Forward with an Abundance Mentality
Larry Buie, PharmD, BCOP, FASHP
HOPA President (2021–2022)
Manager, Clinical Pharmacy Practice
PGY2 Residency Program Director, Memorial Sloan Kettering Cancer Center
New York, NY
Our vision is so simple, that all patients with cancer will have an oncology pharmacist as an integral member of their care team. Collectively, we support pharmacy practitioners and promote and advance hematology/oncology pharmacy to optimize the care of individuals affected by cancer. However, achieving our mission and vision are complicated if we don’t approach it with an abundance mentality.
You may ask yourself what is an abundance mentality? For me, it means that we dream big and outside the box. We are focused on moving our organization and our profession forward. This encourages us to plan strategically, to embrace change, and seize opportunities. With an abundance mentality, we move forward with positivity and optimism. We take risks with the belief that we will make mistakes, we will learn from them, and we will be better for it. Together, we are stronger.
An Abundance Mentality Comes to Fruition
Diversity, Equity and Inclusion. We have formed a DEI Task Force to put the spotlight on our DEI initiatives. This group has published a DEI statement and made recommendations to the HOPA Board of Directors that span all of our strategic pillars (professional development, professional resources and tools, research, and advocacy), as well as leadership development, and governance. Perhaps most importantly, the task force helps create a culture where it is safe – and expected – to have conversations about diversity, equity, and inclusion.
Change takes time and we know we have a continued need for a DEI lens moving forward. Therefore the DEI task force will be converted to a standing committee.
Collaborative Research. HOPA’s Oral Chemotherapy Collaborative (OCC) provides a collaborative research framework that promotes oral chemotherapy best practices across all four HOPA councils to improve the quality of care for our patients. To learn more about the OCC and their goals, look for related sessions at our upcoming Annual Conference and be on the lookout for additional opportunities with the ASCO Quality Training Program.
Amplifying the Patient Voice. This year we created HOPA’s first Patient Advisory Panel. The panel is charged with providing the patient perspective across all HOPA initiatives and we were fortunate to have panel participation at our fall Hill Day. HOPA is also currently redesigning our website for an enhanced patient experience.
Expanding our Reach. At HOPA, we believe that we should be the professional home to all pharmacists that are taking care of oncology patients. This begins by engaging our student members and teaching them about all that HOPA has to offer. Our student engagement task force was formed and recommended that a National Student Group Committee be created. We are so excited to bring the student voice to HOPA!
Mitigating Pharmacist Burnout. At last year’s annual conference Allison Golbach’s research revealed that >60% of oncology pharmacists were experiencing burnout. The study also detailed consequences of burnout, including increased risk of making medication errors. HOPA is dedicated to working towards solutions that will support our membership and has engaged a consultant to help us identify ways to support pharmacists experiencing burnout and to prevent burnout where we can.
Annual Conference: A HOPA Reunion in Boston!
A year has already come and gone and by the time you see this in print, I may be introducing your new HOPA President, Heidi Finnes, at our in-person annual conference in Boston! It is hard to believe that it has been nearly two years since we have been able to be together in person. The lineup of science and networking is too great for me to preview in full in this letter but here are a few anticipated highlights:
- John G. Kuhn Keynote Address “Bite-Sized Well-Being During Times of Uncertainty,” presented by J. Bryan Sexton, PhD will address healthcare worker burnout and evidence-based approaches to reduce it.
- The session, “Optimizing Care for Patients Taking Oral Anti-Cancer Agents: How HOPA’s OCC is Moving Practice Forward” will demonstrate the OCC’s collaborative framework.
- The first-ever HOPA DEI Award will be presented to Maurice Alexander and Britny Brown, the Chair and Vice-Chair of HOPA’s DEI Task Force.
I have been privileged to serve as President of HOPA, and I began and will end this journey with gratitude. Gratitude for the amazing care you provide our cancer patients. Gratitude for the opportunity to work alongside an amazing Board of Directors. Gratitude for our volunteers - moving our organization forward with an abundance mentality. We are all winning together!
Feature: B-Cell Maturation Antigen Targeting CAR T-Cell Therapy in Relapsed/Refractory Multiple Myeloma
Abigail Shockley, PharmD
PGY2 Hematology/Oncology Pharmacy Resident
Medical University of South Carolina
Charleston, SC
James Davis, PharmD, BCOP
Assistant Professor – MUSC College of Pharmacy
Malignant Hematology Clinical Pharmacy Specialist
Medical University of South Carolina
Charleston, SC
Background
Multiple myeloma is characterized by malignant proliferation of plasma cells in the bone marrow.1-4 These abnormal plasma cells overproduce immunoglobulins causing protein accumulation, bone marrow failure, bone destruction, and end-organ damage.1 Multiple myeloma is the second most common hematologic cancer and accounts for 1.8% of all cancers with an estimated 32,000 new cases in the United States in 2020, and more than 12,000 deaths.2-4 Although many patients obtain deep and durable remissions with induction therapy, relapse is inevitable in this incurable disease.
There are many available treatments for relapsed/refractory (R/R) disease but ideal sequencing remains a challenge. The National Comprehensive Cancer Network (NCCN) guidelines contain nine different preferred category 1 treatment regimens for these patients.4 These treatment combinations consist of multiple agents including: antiCD38 monoclonal antibodies, proteasome inhibitors, immunomodulatory agents, and corticosteroids. With each relapse and subsequent treatment, progression-free survival (PFS) and overall survival (OS) outcomes become shorter.4,5 The poor prognoses of these patients have led researchers to investigate novel treatment modalities, including chimeric antigen receptor (CAR) T-cell therapy.
CAR T-cell therapy is a type of treatment in which a patient’s T cells are genetically modified in a laboratory to express a CAR that is specifically designed to target select cancer cells’ surface markers. This process is initiated by collecting T-cells from the patient’s blood via leukapheresis. The collected T-cells are then sent to a manufacturing lab for CAR attachment and proliferation. This process generally takes between 4 to 6 weeks. Once the T-cells meet manufacturing specification, the cells are shipped back to the medical center for infusion into the patient. Prior to cell infusion, the patient undergoes lymphodepleting chemotherapy in order to make a favorable environment for T-cell expansion and persistence. After the infusion, the CAR T-cells target, attack, and destroy the cancer.1
B-cell maturation antigen (BCMA) is a cell surface protein that is a member of the tumor necrosis factor (TNF) receptor family primary expressed on plasma cells. BCMA regulates the maturation of B-cells into plasma cells. Multiple myeloma cells have been shown to overexpress BCMA which leads to malignant plasma cell survival.6 BCMA is also undetectable in naïve B-cells, hematopoietic stem cells, and normal non-hematologic tissues. This unique expression of BCMA on myeloma cells has allowed researchers to develop BCMA targeted therapies for myeloma while being able to reduce off-target toxicities.1,6
Recently, efficacy and safety outcomes of two BCMA targeting CAR T-cell therapies, idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cilta-cel) were published.7,8 Here, we summarize and discuss the literature for these novel BCMA targeting CAR T-cell therapies.
KarMMa: Idecabtagene Vicleucel
KarMMa was an open-label, multicenter, phase 2 trial that sought to confirm the safety and efficacy of idecabtagene vicleucel in patients with R/R multiple myeloma. One hundred twenty-eight patients aged 33-78 years (median 61) received one infusion of ide-cel at a target dose of 150x106, 300x106, or 450x106 per kilogram CAR-positive T-cells 2 days after receiving lymphodepleting chemotherapy. The median lines of therapy prior to study enrollment were 6 (3-16). Sixteen percent of patients had Revised–International Staging System (R-ISS) stage III disease and 35% had high-risk cytogenetic abnormalities defined as del(17p), t(4;14), or t(14;16). Patients were excluded if they had evidence of plasma cell leukemia, previous allogeneic hematopoietic stem cell transplantation, and/or central nervous system disease.7
The primary endpoint was overall response rate (ORR). The secondary endpoint was complete response (CR) or stringent CR (sCR) rates. At a median follow-up of 13.3 months, 73% of patients met the primary outcome of ORR (95% CI 66-81; p<0.001) with 33% obtaining a CR or better. Twenty-six percent of the patients achieved minimal residual disease (MRD) negative status. The median duration of response (DOR) was 10.7 months (95% CI 9.0- 11.3). Median PFS and OS were 8.8 months (95% CI 5.6-11.6) and 19.4 months (95% CI 18.2-could not be estimated), respectively.7
Treatment related toxicity was reported in all patients receiving treatment with 99% of patients experiencing a grade ≥3 adverse reaction. The most common high-grade toxicities were neutropenia (89%), anemia (60%), thrombocytopenia (52%), and infections (22%). Other notable toxicities included cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). Eighty-four percent of patients experienced low-grade CRS with 5% of patients experiencing grade ≥3 toxicity. The median time to onset of CRS was 1 day (1-12), with a median duration of 5 days (1-63). Eighteen percent of patients experienced ICANS, with 3% of patients experiencing grade ≥3 neurotoxicity. Median time to onset for ICANS was 2 days (1-10), with a median duration of 3 days (1-26).
On March 26, 2021, the FDA granted idel-cel accelerated approval for adult patients with R/R myeloma after 4 or more lines of therapy, including an immunomodulatory agent, a proteasome 4 inhibitor, and an anti-CD38 antibody, based on the results of KarMMa trial.7 The NCCN also added idecabtagene vicleucel as a category 2A recommendation based on this approval.4,7
CARTITUDE-1: Ciltacabtagene Autoleucel
CARTITUDE-1 is a multicenter, open-label, single arm, phase 1b/2 trial that evaluates dosing, safety, and efficacy of ciltacabtagene autoleucel in patients with R/R multiple myeloma. Ninety-seven patients aged 56-68 (median 61) received cilta-cel at a target dose of 0.75x106 CAR-positive T-cells per kilogram after lymphodepleting chemotherapy. Patients were required to have failed previous treatment with at least 3 lines of therapy with the median lines of therapy prior to study enrollment being 6 (4-8). Fourteen percent of patients had R-ISS stage III disease and 24% had high-risk cytogenetic abnormalities defined as del (17p), t(4;14), or t(14;16). Patients were excluded if they had previous CAR T-cell or BCMA targeting therapy.8
The primary endpoints were safety, dose confirmation, and ORR. Secondary endpoints were CR, sCR, DOR, PFS, MRD-negativity rate, and OS. At a median follow-up of 12.4 months, 97% of patients met the primary outcome of ORR (95% CI 91.2–99.4; p<0.001) with 67% obtaining a sCR. Ninety-three percent of patients achieved MRD negative status. The median DOR (15.9-not estimable) and PFS (16.8-not estimable) were not reached. 12-month OS was 89% (80.2-93.5).8 A 24-month update presented at the 2021 American Society of Hematology Annual Meeting, confirmed these results with ORR in 97.9% of patients with 82.5% maintaining a sCR. 24-month PFS and OS were 60.5% and 74%, respectively.9
Treatment related toxicity was reported in all patients receiving treatment with 99% of patients experiencing a grade ≥3 adverse reaction. The most common high-grade toxicities were neutropenia (95%), anemia (68%), thrombocytopenia (60%), and infections (20%). Ninety-five percent of patients experienced low-grade CRS with 4% of patients experiencing grade ≥3 toxicity. The median time to onset of CRS was 7 days (IQR 5-8), with a median duration of 4 days (IQR 3-6). One patient died due to grade 5 CRS in combination with hemophagocytic lymphohistiocytosis (HLH). Twenty-one percent of patients experienced neurotoxicity with 9% of patients experiencing grade ≥3 events. Median time to onset was 8 days (IQR 6-8), with a median duration of 4 days (IQR 6-8). Eight percent of patients experienced delayed high-grade neurotoxicity not described by ICANS diagnostic criteria. These events included parkinsonian-like movement disorders, cranial nerve paralysis, and neuropathy. Fifty percent of these patients’ neurotoxicity did not resolve and one patient death occurred from a grade five event.8
The manufacturer anticipated a decision from the FDA on the accelerated approval of cilta-cel in the fourth quarter of 2021; however, the FDA’s decision was recently postponed until the first quarter of 2022.10 There are other ongoing trials using cilta-cel in combination with other anti-myeloma therapies as well as in earlier treatment lines for patients with multiple myeloma.
Treatment Considerations:
Advantages
There are many advantages of the utilization of CAR T-cell therapy in R/R myeloma patients. In comparison to other chemotherapy regimens, which require multiple infusions administered at pre-defined intervals, CAR T-cell therapy works with the immune system to provide a deep response after a single infusion. CAR T-cells may provide heavily pre-treated patients a substantial treatment-free interval during remission. For these reasons many patients report to have improved quality of life following ide-cel infusion.11 BCMA targeting CAR T-cell therapies can offer many patients previously thought to have run out of treatment choices the option of prolonged, treatment-free survival.
Toxicity
CAR T-cell therapy is associated with significant toxicities including CRS, neurotoxicity, prolonged cytopenias, and infections.7,8 Due to ide-cel’s CRS and neurotoxicity there is a risk evaluation and mitigation strategy (REMS) program which ensures that hospitals and their associated clinics that dispense this CAR T-cell therapy are certified to manage CRS and neurotoxicity and have access to tocilizumab, a medication approved to manage these toxicities.7 These toxicities also make CAR T-cell therapy challenging to administer safely in frail patients, those with high disease burden, and those who are heavily pretreated. Treatments of these toxicities are improving, however, patients are still required to receive CAR T-cell therapy and monitoring at large tertiary medical centers. An advantage of cilta-cel’s delayed CRS and neurotoxicity may potentially allow for outpatient administration and better reimbursement, though, these delayed toxicities may pose a challenge to successful long-term follow-up. Many ongoing studies are working to overcome CAR T-cell toxicity through modifications of CAR T-cell constructs. Methods to mitigate toxicity include utilizing suicide switch mechanisms, dual target antigen recognition, synthetic notch receptors, inhibitory CARs, bispecific T-cell engagers, and more.12
Therapeutic Resistance and Duration of Response
The limited duration of responses to CAR T-cell therapy suggests that current constructs may be vulnerable to resistance. The mechanisms behind resistance and therapeutic failure are poorly understood.13 Ide-cel’s short persistence is thought to be due to the lack of ability to produce a robust response by memory T-cells. In ongoing trials, ide-cel’s CAR construct is being studied with the addition of a phosphoinositide 3-kinase inhibitor during the CAR T-cell expansion phase. This is hypothesized to enhance the drug product’s memory in hopes to provide a more durable response.14
Cilta-cel’s increased durability may be due to improved binding affinity. Cilta-cel expresses two BCMA-targeting single-domain antibodies and a CD3-41BB co-stimulatory domain to optimize T-cell activation and proliferation.8 Additionally, future research to mitigate antigen escape is currently underway. Antigen escape can occur as CAR T-cells are targeting a specific antigen, thus applying selective pressure on malignant clones that do not express that target. This ultimately results in disease relapse due to the replication of the surviving clones. It is thought that this mechanism of resistance can be mitigated through dual targeting via selection of multiple antigens.15
Accessibility
Access to BCMA targeting CAR T-cell products remains limited. At this time, ide-cel is the only currently FDA approved CAR T-cell therapy for multiple myeloma and manufacturing slots have not been able to keep up with prescriber demand. Cilta-cel approval has been delayed and roll-out may take months due to the anticipated REMS requirements.10 Additionally, patients must meet many requirements prior to receiving therapy. Patients must have good social support, access to a tertiary medical center for monitoring, be fit enough to make it through the 4-to-6-week manufacturing process, and have insurance approval. The costs of these therapies are substantial for both the institution and patient. It is estimated that the average cost for CAR T-cell therapy is approximately 700,000 dollars and can increase to over 2 million dollars depending on the extent of monitoring and treatment of toxicities.16
Cilta-cel’s delayed toxicity profile may potentially allow outpatient administration, thereby increasing access and potentially increasing insurance reimbursement. Allogeneic or “off the shelf” CAR T-cell products are also under investigation and may improve product-to-vein time, eliminating the need for leukapheresis, and potentially lowering costs.13,17 Ongoing BCMA targeting CAR T-cell therapies can be seen in Table 1.
Conclusion
CAR T-cell therapy has revolutionized the treatment of R/R multiple myeloma. Both the KarMMa and CARTITUDE-1 trials showed improved survival outcomes in this difficult to treat patient population. Ide-cel has also demonstrated increased quality of life. Among these BCMA targeting CAR T-cell therapies, cilta-cel seems to have the highest response rates; however, caution should be used when comparing results between trials. These novel cellular therapies provide heavily pre-treated patients with promising options. As data mature and these therapies become more accessible, BCMA targeting CAR T-cell therapy will likely become more frequently incorporated into the multiple myeloma treatment paradigm.
Table 1. Ongoing Trials of BCMA Targeting CAR T-cell Therapy
| Agent | Clinicaltrials.gov identifier (NCT #) | Phase | Patient Population and treatment | Purpose | Outcome |
|---|---|---|---|---|---|
| bb21217 | NCT03274219 | I | R/R MM | New CAR construct to enhance binding affinity | Safety and efficacy |
| CXCR4 modified anti-BCMA CAR T cells | NCT04727008 | I | R/R MM | Improve infiltration of human natural killer cells into the bone marrow | Dosing, safety, and tolerability |
| ALLO-715 | NCT04093596 ‘UNIVERSAL’ |
I | R/R MM | Allogeneic CAR T cells to improve accessibility | Safety, efficacy, cell kinetics, and immunogenicity |
| Dual specificity CD38 and BCMA | NCT03767751 | I/II | R/R MM | Dual targeting | Dosing, safety, cell kinetics, efficacy |
| Dual specificity CD19 and BCMA | NCT04714827 | II | R/R MM | Dual targeting | Dosing, safety, cell kinetics, efficacy |
| LCAR-B38M | NCT03758417 | II | R/R MM | Dual epitope binding on BCMA | Kinetics, efficacy, safety |
| JNJ-68284528 | NCT04133639 | II | Consolidation | Replace ASCT in front-line therapy | Kinetics, efficacy, safety |
| ALLO-647 | NCT05000450 | II | R/R MM | Allogeneic CAR T cells to improve accessibility | Safety, efficacy, cell kinetics, and immunogenicity |
| CT053 | NCT03915184 ‘LUMMICAR-2’ |
I/II | R/R MM | 8-to-10-day manufacture time | Safety and efficacy |
| Descartes 11 | NCT03994705 | I/II | R/R MM | MRNA engineered to eliminate preconditioning | Dosing, safety, and activity |
| NCT04436029 | II | Consolidation | MRD and efficacy post induction |
ASCT: autologous stem cell transplant; CAR: chimeric antigen receptor; MM: multiple myeloma; R/R: relapsed/refractory; MRD: minimal residual disease.
REFERENCES
- Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046- 1060. doi:10.1056/NEJMra1011442
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. CA: A Cancer Journal for Clinicians 2021; 71: 7–33
- Myeloma - Cancer Stat Facts. SEER, https://seer.cancer.gov/statfacts/html/mulmy.html (Accessed 1 October 2021)
- National Comprehensive Cancer Network. Multiple myeloma (version 4.2022), www.nccn.org/professionals/physician_gls/pdf/myeloma.pdf (accessed 27 December 2021)
- Kumar SK, Dimopoulos MA, Kastritis E, et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: a multicenter IMWG study. Leukemia. 2017;31(11):2443-2448. doi:10.1038/leu.2017.138
- Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048-2060. doi:10.1158/1078-0432. CCR-12-2422
- Munshi NC, Anderson LD Jr, Shah N, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med. 2021;384(8):705- 716. doi:10.1056/NEJMoa2024850
- Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study [published correction appears in Lancet. 2021 Oct 2;398(10307):1216]. Lancet. 2021;398(10297):314-324. doi:10.1016/S0140- 6736(21)00933-8
- Martin T, Usmani SZ, Berdeja JG, et al. 549 Updated results from CARTITUDE-1: Phase1b/2 study of ciltacabtagene autoleucel, a b-cell maturation antigen chimeric antigen receptor t cell therapy, in patients with relapsed/refractory multiple myeloma. ASH update 12 December 2021. https://ash.confex.com/ash/2021/webprogram/Paper146060.html (accessed 14 December 2021)
- Young J, Chen C. Legend biotech announces extension of PDUFA date for cilta-cel. Legend Biotech. https://investors.legendbiotech.com/news-releases/news-release-details/legend-biotech-announces-extension-pdufa-date-cilta-cel (accessed 27 December 2021)
- Shah N, Delforge M, San-Miguel JF, et al. 436 secondary quality-of-life domains in patients with relapsed and refractory multiple myeloma treated with BCMA-directed CAR T cell therapy. Presented at ASH 2020. December 6, 2020. https://ash.confex.com/ash/2020/webprogram/Paper136665.html (accessed 12 December 2021)
- Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. 2019;18(1):125. Published 2019 Aug 20. doi:10.1186/s12943-019-1057-4
- Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16(6):372-385. doi:10.1038/s41571-019-0184-6
- Berdeja JG, Alsina M, Shah ND, et al. Updated Results from an Ongoing Phase 1 Clinical Study of bb21217 Anti-Bcma CAR T Cell therapy. Blood. 2019 Nov 13;134(S1):927
- Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. doi:10.1126/ scitranslmed.3006597
- Prime Therapeutics’ study shows total cost of care for CAR-T plus post-treatment events can exceed $1 million [news release]. Prime Therapeutics; 12 April, 2021. https://www.primetherapeutics.com/en/news/pressreleases/2021/release-2021-cost-of-care-car-t-exceed-million.html (Accessed 13 December 2021)
- Mailankody S, Matous JV, Liedtke M, et al. An Allogeneic First-in-Human Study of the Anti-Bcma ALLO-715 and the Anti-CD52 ALLO-647 in Relapsed/Refractory Multiple Myeloma. Oral presentation/abstract presented at the American Society of Hematology annual meeting. 2020 Dec 5
Navigating the Pandemic: Academic Prospective, What is Next?
Nelly G. Adel, PharmD, BCOP, BCPS
Chair Pharmacy Practice,
Associate Professor, Oncology
Touro College of Pharmacy
New York, NY
In March 2020 we were all hit by the reality of moving to a virtual presence. With the support of the college, everyone was obligated to get the necessary training to be able to upload, record, and deliver a meaningful lecture virtually. The questions remained: Can faculty deliver Problem Based Learning (PBL) or Team Based Learning (TBL) virtually without having the ample time to prepare for it? Can faculty deliver an effective lecture without seeing students on camera and ensuring students are truly present and participating?
The challenge was even harder for pharmacy students as they sought availability of computers and internet access; balanced family responsibilities, house situations, various obligations, and college work; and still had to be present for synchronized teaching. It was almost impossible to keep up with the continuous changes and demands. Many students went through tough times, losing family members, and were subjected to unforeseen hardships.
More Questions than Answers
Another challenge in academia was the assessment. We wondered how to assess the students’ performance while they were home with access to their learning resources during exams. Fortunately, many examination software programs have implemented new ways to proctor students while they take exams at home. This by itself added a tremendous element of training for both faculty and students that added to the workload on both sides.
The Office of Pharmacy Experience (OPE) was not exempt from this huge crisis. How can OPE create various experiences for students to graduate on time? How can OPE create Ambulatory Care experiences utilizing Telehealth, taking into consideration the Health Insurance Portability and Accountability Act (HIPPA) requirements and student access to medical records “virtually”? Retail pharmacies were sending students home and hospital sites refused to accept any students for a long time period (more than one advanced pharmacy practice experience; APPE.) Families were concerned about the health of their students going to sites that were loaded with COVID-19 cases. It was a true nightmare with huge disappointments for many students who were awaiting their graduation.
Surviving Early Challenges
During the early time of the pandemic, the Accreditation Council for Pharmacy Education (ACPE) began to set expectations for virtual rotations and provide guidance to pharmacy schools.
In the northeast, as we awaited ACPE guidance, the pandemic hit states at various points, and the timeline was different for all of us. When New York State was in the peak of the pandemic other pharmacy schools in the country were not yet affected. By the time New York was out of risk, other states started to shut down.
The guidance delivered by ACPE was not timely to all of us; individuals needed to tailor their programs, which led to creativity in delivering experiential education without consistency. For many of us, having to experience the virtual world brought a new perspective to our careers, workload, and work environment; we had a sense that there is more to life than just work. Many pharmacists are reconsidering new opportunities or finding new jobs with more flexible hours. Faculty are leaving colleges, retail pharmacists are looking for other opportunities, and clinical pharmacists are leaving into early retirement.
Improving the Future
During the American Society of Health-System Pharmacists Midyear Clinical Meeting (ASHP-MCM) in 2019, there was a meeting with all the Deans of pharmacy schools to address the decreased number of applicants. At that time, the impression was that pharmacists in general were not satisfied or impressed by their work, the burnout was intolerable. Imagine now, two years later, with more and more responsibilities and more challenges. Is there still going to be a good number of applicants to have a better selection process? Or are we stuck with a limited number of well-prepared student applicants?
We have been in this situation for almost two years. What is puzzling is the fact that we human beings with all these advancements, cannot control a virus! This is the reality, no denial there. We are still in the middle of the pandemic with the COVID-19 omicron variant threatening to hit the US with its high contagious rates and fast spread, we will continue to face other new challenges.
The questions for the New Year 2022, are many: are classes and schools going to go virtual for a short time until things are in better control? Are we going to be wearing masks for a longer time? Are the experiential education and expectations of pharmacy schools and clinical experiences going to remain the same? Can pharmacists hold their jobs for a little bit longer with more vaccine doses to be administered and more prescriptions for new oral therapies to be filled?
I want to end with a positive note, the only way to have a better control is to adapt to this reality, we must create new ways to deliver better education and accept that old ways cannot be replicated. We live in a new world and we should be up to this challenge. We can do better.
Integration of a Specialty Pharmacy Team within Oncology Care
Kristina Lo, PharmD, BCOP
Senior Clinical Pharmacist, Oncology Specialty Pharmacy
UC Davis Health
Sacramento, CA
Iris C. Zhao, PharmD, BCPS, BCOP, APh
Senior Clinical Pharmacist, Oncology Specialty Pharmacy
UC Davis Health
Sacramento, CA
Introduction
Oral oncolytic development has been increasing steadily, making up about 25-35% of the new oncology drug pipeline.1 With this shift towards oral oncolytic use, many parts of the healthcare system need to evolve to match the pace of this change. While physicians, allied health professionals, and other medical personnel try to keep up with these changes, one of the greatest challenges that plague healthcare systems is staff shortages across all disciplines.2 Timely care, medication education, and medication access are just a few of the many things that can be negatively affected by staff shortages, potentially leading to poorer clinical outcomes.3 To help bridge these gaps, we developed the Oncology Specialty Pharmacy (OSP) at UC Davis in the adult hematology/oncology clinic setting with a focus on oral oncolytics.
Initial Integration into the Hematology/Oncology Clinic
Our OSP started as an initial year-long pilot in 2013 with one pharmacist providing telephone calls to patients one day a week to monitor adherence and toxicities. In September 2014, 1 full-time employee (FTE) pharmacist and 1 FTE technician were introduced into the adult hematology/oncology clinic setting to work directly with our multi-disciplinary staff. Since then, we have expanded to 4 FTE pharmacists and 4 FTE technicians over the last 7 years.
The OSP and clinic are staffed by clinical pharmacists with appropriate experience and training, such as completion of a PGY1 Pharmacy Residency Program with an emphasis in Oncology and/or a PGY2 Oncology Pharmacy Residency Program, or equivalent (e.g., Board Certified Oncology Pharmacist). Experience with the hematology/oncology patient population and demonstrated competence in cancer therapy and supportive care medication management are necessary for pharmacists prior to involvement in the OSP.
There is no specific requirement for technicians who staff the OSP and clinic, though experience with financial assistance and prior authorizations (PAs) is preferred. Currently at UC Davis all technicians have at least 5 years of experience in various settings from retail and/or specialty settings. The pharmacy team plays a pivotal role in relieving significant burden from patients experiencing the financial toxicities of their therapies through applying for financial assistance on behalf of the patient. In addition, having dedicated pharmacy technicians to submit PAs can decrease the time to initiation of therapy.
Clinical Pharmacy Services – A Hybrid Model
Outpatient pharmacy service models across the nation vary in the degree of integration into the clinic setting. Traditionally, specialty pharmacies have limited or no affiliation with the patient’s practice-based oncology care team. This can lead to significant logistical barriers to oncology care including a lack of shared health records for medical communication and documentation.4
The integrated OSP service at UC Davis allows the oncology pharmacists and technicians a unique opportunity to serve as both a tele-provider to patients via phone-based services and on-site support to providers and patients in the clinic. While our initial pilot program only monitored adherence and toxicities, our list of services was later expanded to meet the most critical needs for our patients once full-time staff were integrated into the clinic space in 2014. Currently, the OSP team provides a full spectrum of services, including clinical evaluation of therapy, PA support, financial assistance, patient and caregiver education, regular lab monitoring, drug/herb interaction evaluation, and supportive care management.
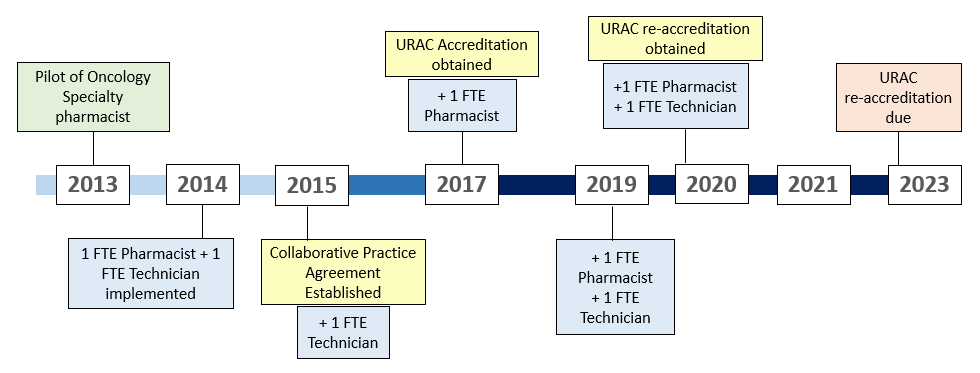
Figure 1: Timeline of the UC Davis Oncology Specialty Pharmacy
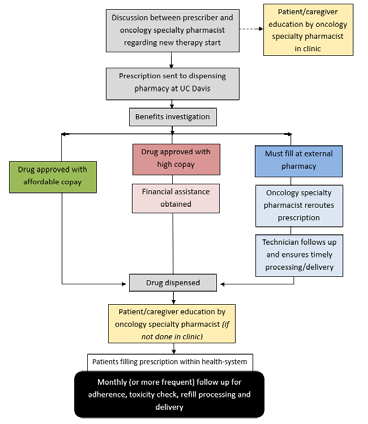
Figure 2: Overview of Oncology Specialty Pharmacy workflow
Initial Prescription of Therapy
Oncology specialty pharmacists play a key role in the discussion of the appropriateness of a therapy and collaborate with providers in determining a dosing and monitoring plan. Once a treatment plan with an oral oncolytic has been established, providers send prescriptions directly to the on-site dispensing pharmacy. The dispensing pharmacy staff are trained to communicate directly with the OSP team in the benefits investigation process and triage prescriptions to dedicated PA oncology technicians who submit PA’s for all cancer center prescriptions. Once the PA process is complete, OSP technicians evaluate prescription copays and drive the patient assistance process for prescriptions with high copays. Patients who are referred to the OSP are added to a confidential list of patients to be educated and monitored by the oncology specialty pharmacists.
Education
In order to make informed decisions about treatment, patients and caregivers must receive education on administration and monitoring of the oral oncolytic, expected outcomes, adverse effects, and costs of therapy.5 Specialty pharmacists provide comprehensive medication education and a full medication reconciliation to all patients who are initiated on an oral oncolytic and ensure pertinent baseline laboratory testing is completed prior to initiation of therapy, if applicable. These actions can be completed over the phone, in-clinic at the time of prescription, or at the dispensing pharmacy at time of dispense.
Clinically Meaningful Management and Follow-up
Once patients initiate therapy, pharmacists follow a standardized monthly interval for pharmacist follow up. Lab tests and recent scans are evaluated to determine continued appropriateness of therapy; patients are assessed for correct administration of medication, adherence, and adverse events; and refills are processed and arranged for pick up or delivery. Evaluation and adjustment of monitoring plans, clinic visits, and/or prescription of additional supportive care prescriptions are also performed at this time. More frequent follow up is implemented for patients that are determined to be at high-risk for non-adherence or adverse events.
Current practice at UC Davis is to conduct education for all patients prescribed an oral oncolytic, whereas regular follow up calls are conducted for patients whose specialty medications are filled at the UC Davis dispensing pharmacy, instead of an external specialty pharmacy. All patients, regardless of their filling pharmacy, are educated to call the OSP team for questions or assistance with adverse event management.
Patient and Provider Impact
To evaluate current practices and services, patients and providers are sent satisfaction surveys to evaluate their interactions with the OSP team. Surveys are created by the National Association of Specialty Pharmacy (NASP) and distributed through either direct mail or web-based options quarterly. These survey results provide the OSP team insight into ways to develop and improve services.
Adherence data
In addition to patient and provider satisfaction, the most important detail is improved patient care and outcomes. While clinical outcomes may be difficult to ascertain, patient adherence is necessary to optimize their chance for a successful outcome. In 2015, a retrospective analysis was performed to evaluate adherence of our OSP and it was observed that our program was able to significantly improve patient adherence.6 Adherence was analyzed through medication possession ratio (MPR).
Challenges and Limitations
Given the recent growth in the use of oral oncolytics and their high-cost nature, more payors are setting limitations and/or mandating which pharmacies a covered patient can receive their specialty medications from. In addition, manufacturers are narrowing the distribution of their specialty medications to certain pharmacies to reduce distribution costs, among other reasons. Payor lockouts and limited distribution models limit the specialty pharmacists’ assessment of adherence and lead to delays in treatment initiation or continuation.7 In order to offset this challenge, the UC Davis OSP team implemented the pharmacy technician service of contacting external specialty pharmacies to follow up on prescriptions rerouted outside of the health system.
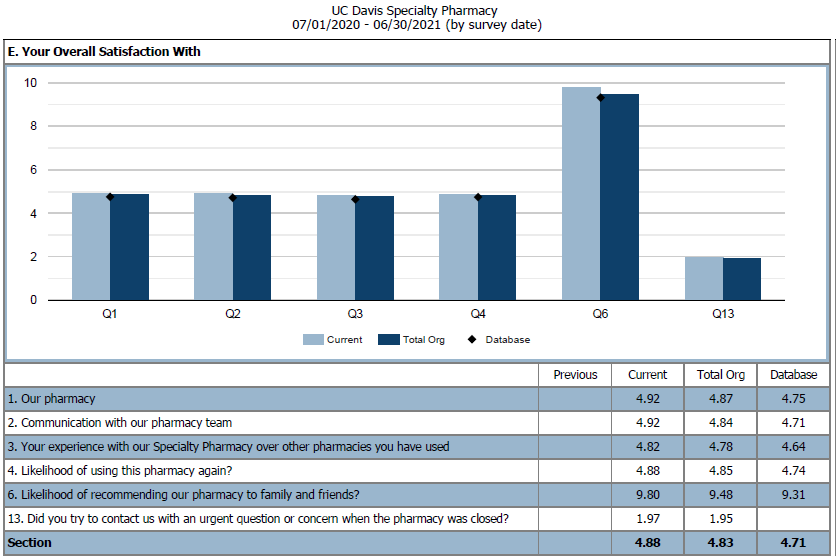
Figure 3: NASP Patient Overall Satisfaction Survey from July 2020 - 2021. We compared UC Davis Oncology Specialty Pharmacy to other UC Davis specialty programs and the NASP database.
Generalized growth in cancer care and use of oral oncolytics has led to an increased need for additional OSP staff which has been challenging to meet from a hiring perspective. Growth in the specialty pharmacy team as well as staff in other interdisciplinary areas had led to a shortage in available clinic space which is critical to continued on-site pharmacist support.
Agreeing upon a single case management program has also been a challenge for the UC Davis OSP. Although there are case management programs available, most programs do not interface with the health systems’ electronic medical record (EMR) which leads to increased time spent on duplicate documentation.
A continual workflow challenge for the OSP team has been ensuring that our practices meet the standards efficiently and thoroughly for multiple accrediting bodies. The UC Davis Cancer Center has been American Society of Clinical Oncology (ASCO) Quality Oncology Practice Initiative (QOPI) certified since 2016 and our specialty pharmacy has been Utilization Review Accreditation Commission (URAC) accredited since March 2017. With both of these accrediting bodies requiring different information, follow-up frequency and documentation are tailored to meet the varying standards from both QOPI and URAC.
Future Directions
There is a continued need for pharmacist outreach to encourage patient adherence to medications and manage supportive care to overcome the clinical implications of payor lockouts and limited distribution models. A primary goal of the UC Davis OSP is to have regular pharmacist outreach for all patients regardless of their filling pharmacy.
The increased use of oral oncolytics and the paralleling growth in challenges provide a glimpse at future opportunities for oncology pharmacists. Currently at UC Davis, specialty pharmacists are caring for any diagnoses and their associated oral therapies. However, there is the potential for pharmacists to become disease state specific which may provide more consistency for providers, nursing, and patients.
Another consideration for the future includes transitioning oral oncolytic orders to EMR oncology treatment modules leading to a more pharmacist-driven prescription process. Challenges include adjusting the specialty pharmacy workflow, provider buy-in and training, and additional information technology (IT) and pharmacist support. Additional IT and pharmacist support is critical as there are numerous oral therapies and their associated monitoring needs are unique to each individual medication.
Conclusion
The oncology pharmacist is a commonly untapped resource that can be utilized to help bridge many gaps that exist in our current healthcare situation. While OSP is simply one way to better optimize the oncology pharmacy staff, this is certainly not without its own challenges. Resources continue to be scarce, with drug and staff shortages rampant worldwide. Despite this, we have shown that OSPs can continue to be an opportunity for institutions to provide the desperately needed relief for both patients and medical staff, while providing the recognition that the pharmacy team can provide so much more than traditionally thought.
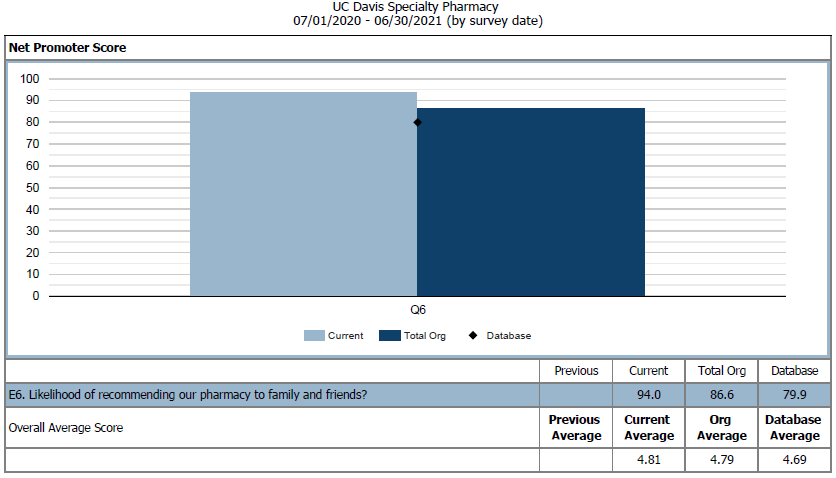
Figure 4: NASP Provider Net Promoter Score for Q6 in 2020 – 2021. We compared UC Davis Oncology Specialty Pharmacy to other UC Davis specialty programs and the NASP database.
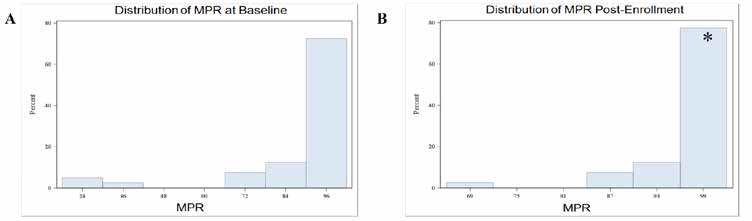
Figure 5: OSP Adherence Analysis from 2013-2014. A. MPR Analysis at Baseline. Mean MPR at baseline was 90.3%. B. MPR Analysis Post-Enrollment into our OSP. Mean MPR post-enrollment was 99.3%. *Mean increase = 9.08% (P=0.02).
REFERENCES
- Stokes M, Reyes C, Xia Y, Alas V, Goertz HP, Boulanger L. Impact of pharmacy channel on adherence to oral oncolytics. BMC Health Serv Res. 2017;17(1):414. doi:10.1186/s12913-017-2373-2
- Solutions to the Oncology Workforce Shortage. Ensuring Quality Cancer Care through the Oncology Workforce: Sustaining Care in the 21st Century: Workshop Summary. Institute of Medicine (US) National Cancer Policy Forum. Washington (DC): National Academies Press (US); 2009. (https://www.ncbi.nlm.nih.gov/books/NBK215257/)
- Lafferty, M et al. Causes and Consequences of Chemotherapy Delays in Ambulatory Oncology Practices: A Multisite Qualitative Study. ONF. 2020, 47(4), 417-427
- Muluneh B, Schneider M, Faso A, Amerine L, Daniels R, Crisp B, et al. Improved Adherence Rates and Clinical Outcomes of an Integrated, Closed-Loop, Pharmacist-Led, Oral Chemotherapy Management Program. J Oncol Pract. 2018 Jun;14(6): e324-e334. doi: 10.1200/JOP.17.00039
- Mackler E, Segal E, Muluneh B, Jeffers K, Carmichael J. 2018 Hematology/ Oncology Pharmacist Association Best Practices for the Management of Oral Oncolytic Therapy: Pharmacy Practice Standard. J Oncol Pract. 2019 Apr;15(4):e346-e355. doi: 10.1200/JOP.18.00581
- Chen Zhao I, Lai J, Seiki B, Wilson M, Brennan L, Li T, Wun T. Evaluation of a pharmacist-driven oral chemotherapy adherence program. Journal of Clinical Oncology. 2015. 33:15_suppl, 6569-6569
- Savage S, Bates J, Muluneh B. Challenges Continue for True Patient- Centered Access. Specialty Pharmacy Times. 2016 June; 7(3)
Pharmacist Contributions to Quality Improvement in Oncology Care Presented in the ASCO Quality Care Symposium 2021
Rachel L. McDevitt, PharmD, BCOP
Clinical Pharmacist Specialist, Ambulatory Oncology
University of Michigan Rogel Cancer Center
Ann Arbor, MI
Gena Hoefs, PharmD
PGY1 Pharmacy Resident
M Health Fairview-University of Minnesota Medical Center
Minneapolis, MN
Elizabeth Graver
PharmD Candidate - Class of 2022
University of Texas at Austin, College of Pharmacy
Austin, TX
Introduction
The fall 2021 American Society of Clinical Oncology (ASCO) Quality Care Symposium showcased many projects and research endeavors aimed at improving the quality and safety of cancer care, including the work of many oncology pharmacists. Three pharmacist-led abstracts aimed at measuring and improving quality care for patients with malignancies are highlighted here.
Improvement in Time to Oral Anticancer Agent Follow-up1
Johengen and colleagues evaluated the time to first follow-up visit for patients initiated on oral anticancer agents before and after implementation of a dedicated oncology pharmacist. Prior to oncology pharmacist implementation, care managers were responsible for follow up in patients prescribed new oral anticancer agents. This group compared the time to first follow-up for 79 patients started on oral anticancer therapies in the pre-pharmacist time period (11/1/20 - 2/28/21) to the follow-up times for 40 patients started on oral anticancer therapies in the post-pharmacist implementation time period (3/1/21 - 4/30/21).
The authors found the median time to first follow-up after oral anticancer therapy initiation was 8 days for patients in the pre-pharmacist group compared to 7 days in the pharmacist group. Additionally, time to first follow-up was within 10 days of initiating an oral anticancer therapy for 67.1% of patients in the pre-pharmacist group compared to 95% of patients in the pharmacist group (p<0.001).
The authors concluded time to first follow-up for patients started on oral anticancer therapy was shorter for patients cared for by a pharmacist. Future directions include characterizing the interventions made by the pharmacist at time of follow-up, evaluating patients’ duration of oral anticancer therapy, and rate unplanned admissions in these patients.
Implementation of an EHR-embedded Decision Support Tool in Community Oncology Practices.2
Flatiron Health, Inc, is a healthcare information technology company that specializes in creating and refining clinical decision support (CDS) tools based on real-world data derived from electronic health records and guideline-recommended treatment regimens.3 In effort to evaluate the prevalence of use of the EHR-embedded CDS tool, Flatiron Assist, investigators tracked how often providers were using the CDS tool to guide treatment decisions for patients diagnosed with non-small cell lung cancer (NSCLC) at 2 months, 6 months, and 1 year after adoption. The rationale for implementing the CDS tool is to document and report various quality metrics in hopes of improving the delivery of value-based cancer care.
Their study found over the 1-year observation period, frequency of use of Flatiron Assist increased from 30% at 2 months to 80% at 1-year. Additionally, the study investigated concordance of NSCLC orders with National Comprehensive Cancer Network (NCCN) Guidelines and NCCN Preferred Treatment regimens, and found that over the observed time period, concordance decreased from 94% at 2 months to 85% at 1 year.
The most common reasons providers reported for ordering non-concordant regimens were physician’s choice, patient status, and newly published evidence. The investigators postulated that the decrease in NCCN-concordant orders was attributed to increased usage or greater comfort with the CDS tool over time.
Based on this data, the investigators concluded that Flatiron Assist was quickly adopted by most prescribers and used for the majority of NSCLC orders over the 1-year observation period, most of which were in concordance with NCCN recommendations. Further research is required to further define workflow time, predictors of non-use and non-concordance orders, and correlation of Flatiron Assist with improved clinical outcomes.
Real-world EGFR Testing Patterns Among U.S. Patients with Advanced NSCLC4
Multiple targetable mutations, including mutations in the epidermal growth factor receptor (EGFR), have been identified in patients with NSCLC and help guide therapy selection. NCCN Guidelines recommend molecular testing for these mutations prior to initiating therapy and after disease progression. However, molecular testing presents its own set of challenges including wait times, access to testing, and cost.5 To better characterize real-world use of EGFR testing, Vanderpoel and colleagues performed a retrospective observational study in a cohort of patients with advanced NSCLC from the Flatiron Health database. The study examined a total of 22,726 patients from 2015 to 2020. Overall, 75% of the patients received at least one EGFR test and 15% of those patients tested positive for an EGFR mutation. Of patients who tested positive for an EGFR mutation prior to first line therapy, 36% received a second EGFR test prior to initiating second line therapy.
Notably, the team found the rate of EGFR mutation testing improved over the study period with an 11% increase from 2015 to 2020. Time from sample collection to test results also decreased from 26 days to 16 days for next generation sequencing, and from 17.5 days to 12 days for polymerase chain reaction tests. Identified areas of improvement included increasing the proportion of patients who receive EGFR testing prior to first line therapy and prior to initiating therapies after disease progression.
Conclusion
These projects and many others illustrate the impact of oncology pharmacists in continuing to provide and optimize safe and effective care for patients with cancer. Regional and national presentations and publications by pharmacists in the quality space will continue to show the value of oncology pharmacists.
REFERENCES
- Johengen E, Davidson A, Beekman KW, et al. Improvement in time to oral anticancer agent follow-up. Journal of Clinical Oncology, 2021; 39 (28): suppl 28, abstr 235. 10.1200/JCO.2020.39.28_suppl.235
- Maniago R, Richey SS, DeVincenzo S, et al. Implementation of an EHR-embedded decision support tool in community oncology practices. Journal of Clinical Oncology, 2021; 39 (28): suppl. 28, abstr 274. 10.1200/ JCO.2020.39.28_suppl.274
- About Us. Flatiron Health, Inc. https://flatiron.com/about-us/
- Vanderpoel J, Pericone C, He J, et al. Real-world EGFR testing patterns among U.S. patients with advanced NSCLC. Journal of Clinical Oncology, 2021; 39 (28): suppl 28, abstr 298. 10.1200/JCO.2020.39.28_suppl.298
- Cuppens K, Lodewyckx L, Demedts I, et al. Real-World Treatment Patterns, Epidermal Growth Factor Receptor (EGFR) Testing and Outcomes in EGFR-Mutated Advanced Non-small Cell Lung Cancer Patients in Belgium: Results from the REVEAL Study. Drugs Real World Outcomes. 2021;8(2):141-152. doi:10.1007/s40801-021-00243-w
Asciminib: A Novel TKI for the Treatment of Chronic Myeloid Leukemia
Deeter R. Neumann, PharmD, BCOP
Clinical Oncology Pharmacist – Hematology and Cellular Immunotherapy
Seattle Cancer Care Alliance, UW Medicine
Seattle, WA
The treatment for chronic myeloid leukemia (CML) was transformed in 2001 when the tyrosine kinase inhibitor (TKI), imatinib, was approved by the United States Food and Drug Administration (FDA).1 Today, TKIs on the market demonstrate the ability to reduce a patient’s leukemic burden and restore their life expectancy back to normal.2-6 However, limitations to TKI therapy still exist and are marked by drug toxicity, drug resistance, or both.7
The recent approval of asciminib by the FDA provides a promising option for patients that have developed either toxicity to or resistance against earlier lines of TKI treatment.8
Asciminib Pharmacology
Prior to the development of asciminib, all marketed TKIs for CML exerted their pharmacologic activity via binding to the catalytic adenosine triphosphate (ATP) site in ABL1.7 Mutations to the ABL1 ATP catalytic site result in the inability of TKIs to modulate BCR-ABL1 activity, thus leading to the loss of TKI response and disease progression. One mutation, T315I, is particularly problematic in that it confers resistance to all TKIs used for the treatment of CML except ponatinib.9 Unlike ponatinib and the other TKIs indicated for CML, asciminib exhibits a novel mechanism of action, allowing it to circumvent all ATP catalytic site mutations. Asciminib accomplishes this feat by acting as a selective allosteric inhibitor of BCR-ABL1 kinase activity.
In its wild-type structure, ABL1 displays autoinhibition via the binding of an N-terminal myristoyl group to a C-terminal myristoyl pocket on the kinase domain. In the pathogenic formation of the BCR-ABL1 fusion protein, the BCR protein fragment replaces the N-terminal myristoyl group of ABL1. Without the myristoyl group present, the myristoyl pocket is left vacant, and ABL1 loses the capacity to self-regulate kinase activity. This results in BCR-ABL1 being a constitutively active kinase.7 Asciminib is able to bind to the myristoyl pocket and is thus capable of turning off BCR-ABL1 kinase activity.7 As we will see, the dose needed to effectively inhibit BCR-ABL1 is dependent on the presence of the T315I mutation and is reflected in the FDA approved label.10
Dose Finding Study and Pharmacokinetic Analysis of Asciminib
The maximum tolerated dose of asciminib was evaluated in the phase 1 trial reported by Hughes et al.11 In this trial, adult patients with relapsed/refractory chronic-phase (CP) or accelerated-phase CML were enrolled. Enrolled patients must have failed treatment with at least two different TKIs or were unable to tolerate prior TKI therapy. Seventy percent of the patients enrolled had been previously treated with three or more TKIs, and at the time of the report, 33 patients (22%) harbored the T315I mutation.11
Final pharmacokinetic (PK)-pharmacodynamic (PD) modeling demonstrated that appropriate inhibitory concentrations of asciminib would be maintained in 100% of patients without the T315I mutation when asciminib was dosed at 40 mg twice daily.11 For the limited number of patients harboring the T315I mutation, doses greater than 150 mg twice daily were required to achieve a major molecular response (MMR). Based on these findings, asciminib dosed at 40 mg BID and 200 mg BID were selected for further investigation.
Asciminib is a highly protein bound molecule that is predominantly metabolized by the liver via direct glucuronidation (27.9%) and CYP450 oxidation (37.8%). Additionally, biliary secretion via breast cancer resistance protein (BCRP) plays a role in the elimination of asciminib (31.1%). A minimal amount of asciminib is also cleared by the kidneys (2.5% unchanged).12 The half-life of asciminib was determined to be 8 hours, allowing for steady state levels to be achieved by day 3 of dosing.11 The special population PK studies performed by Hoch et al. focused on individuals with renal and hepatic dysfunction.
The study found increases in both exposure and maximum plasma concentrations in individuals with severe renal impairment (absolute glomerular filtration rate (aGFR) < 30 mL/min and not yet requiring dialysis) and individuals with mild (Child-Pugh class A) and severe hepatic impairment (Child-Pugh class C).12 While PK parameters were impacted, asciminib’s therapeutic window proved to be large enough to conclude that renal and hepatic impairment have little effect on its safety profile. This led to the FDA approving the label for asciminib with no dose adjustments for patients with kidney or liver dysfunction and prescribed 40 mg BID.12
Liver and kidney dysfunction may not be a major determinant for the metabolism of asciminib; however, drug-drug interactions (DDIs) should be evaluated for each patient. Asciminib is a substrate of CYP3A4, and an inhibitor of CYP3A4, CYP2C9, and P-glycoprotein (P-gp).10 Clinicians should consult the prescribing information for guidance on the management of DDIs with asciminib, as the recommendations vary from close monitoring to the avoidance of concomitant medications depending on the prescribed dose of asciminib.10
Efficacy of Asciminib
The results of two pivotal trials led to the FDA approval of asciminib. The ASCEMBL study was an open-label, active-controlled, multicenter, phase 3 trial comparing asciminib with bosutinib.13 Eligible patients were randomized to receive either asciminib 40 mg twice daily or bosutinib 500 mg once daily, and the study’s primary endpoint was the rate of MMR at 24 weeks.13 At 24 weeks, 25.5% of patients receiving asciminib obtained a MMR compared to only 13.2% of patients receiving bosutinib, and the benefit of asciminib was irrespective of the number of previous lines of TKI therapy.13
Updated efficacy data was recently presented at the 2021 American Society of Hematology (ASH) Annual Meeting.14 At 48 weeks of treatment, 33.2% of patients receiving asciminib achieved MMR compared to 18.6% in those receiving bosutinib.14 While initial results for the ASCEMBL study helped asciminib gain an accelerated approval in patients treated with two or more TKIs, the 48 week update demonstrated continued superiority with asciminib.8,13,14
For individuals with known T315I mutations, the efficacy of asciminib continues to be evaluated in the pivotal study, CABL001X2101.8 CABL001X2101 is an ongoing study that expands on the results of the previously reported dose finding trial.8,11 At the time of the FDA’s review, 45 patients with T315I mutated CP CML who have failed at least one other line of TKI therapy were enrolled. The efficacy analysis evaluated MMR at 24 weeks, of which 42% of patients achieved when treated with asciminib. By week 96 of asciminib treatment, 49% of the patients achieved MMR.8
While the pivotal trials showcase the efficacy of asciminib, it is important to evaluate the safety profile of asciminib as this is an important consideration for the selection of TKI therapy in CML.
Safety of Asciminib
The safety profile of asciminib was closely evaluated in the phase 1 dose finding trial reported by Hughes et al.11 Asciminib demonstrated a profile of low severity adverse events (AEs), with 92% of nonhematologic AEs being grade 1 or 2. The most frequent nonhematologic AEs were asymptomatic increases in the lipase or amylase level, rash, and constitutional symptoms. The most common cardiovascular AE was hypertension reported in 19% of patients. All grade thrombocytopenia was the most commonly reported hematologic AE with 22% of patients, and 9.3% experiencing Grade 3 or 4 thrombocytopenia. All grade anemia and neutropenia were reported in 11.3% and 10.7%, respectively.11 The incidence of thrombocytopenia and neutropenia were higher in the Phase III ASCEMBL study, while anemia was similar.13 Clinical pancreatitis was experienced by five patients in the dose finding study (3 patients at the 80 mg BID dose level, 1 patient at the 150 mg BID dose level, and 1 patient at the 200 mg once daily dose level); however, clinical pancreatitis was not reported in the ASCEMBL study.11,13 The 48- week update to the ASCEMBL trial demonstrates that the safety profile remains similar to the primary analysis originally reported by Réa et al.14 While the updated results indicate that myelosuppression occurs earlier in treatment with asciminib, both thrombocytopenia and neutropenia were the most common AEs leading to treatment discontinuation (3.2% and 2.6%, respectively).14 These AEs are important to consider, particularly when considering dose adjustments.
Dose Adjustments and Toxicity Management
The prescribing information for asciminib provides recommendations for dose adjustments due to AEs. The AEs requiring dose adjustment include neutropenia and/or thrombocytopenia, asymptomatic elevations in amylase or lipase, and all Grade 3 or higher nonhematologic AEs.10 The management of all AEs require holding asciminib until near resolution; however, when managing neutropenia or thrombocytopenia, the time to resolution and if it is recurrent dictate whether a dose reduction is warranted.10
While there were no reports of clinical pancreatitis in the ASCEMBL study, there were five cases documented in the phase 1 dose finding study, and all cases occurred at higher doses.11,13 Trial investigators discontinued asciminib in the setting of clinical pancreatitis, and the cases were noted to have resolved within 10 days. One of the five patients was rechallenged and was able to tolerate continued therapy. The study notes that three of the five patients had experienced pancreatitis with previous TKIs. As such, CML patients harboring the T315I mutation where a higher dose is indicated and who have a history of elevated amylase/lipase and/or clinical pancreatitis to another TKI therapy, may be at greater risk for developing pancreatitis on asciminib.
In addition to monitoring for pancreatitis, the safety analysis performed in the special population PK study conducted by Hoch et al. revealed that patients with severe renal impairment had a higher incidence of increased amylase levels or neutropenia.12 While the FDA approved asciminib’s label without dose modifications in renal or hepatic impairment, it would not be unreasonable to consider a lower initial dose of asciminib and increase the dose as tolerated in patients with extensive renal dysfunction.
Place in Treatment and Future Directions
The current National Comprehensive Cancer Network (NCCN) Guidelines for CML recommend selecting frontline TKI therapy based on “risk score, toxicity profile, patient’s age, ability to tolerate therapy, and the presence of comorbidities.”15 The NCCN recommendations are also based on the lack of an overall survival benefit between imatinib and second-generation TKIs. However, given the ability of the second-generation TKIs to decrease the risk of disease progression, they are preferred for patients with intermediate- or high-risk scores.15 For patients that do not respond to a second-generation TKI or have TKI resistance due to the T315I mutation, ponatinib is an effective option.15,16 While asciminib showed superiority over bosutinib in the ASCEMBL study, further investigation is needed to truly understand its place in the treatment of CML patients with the T315I mutation.
The treatment of CML patients with the T315I mutation would be better understood from a study investigating asciminib versus ponatinib. The investigators of the ASCEMBL study noted ongoing efforts to optimize ponatinib dosing as the reason for not using it in the comparator arm against asciminib.13 However, without this information, the demonstrated safety profile of asciminib in the pivotal studies provides it with an advantage over ponatinib in patients with known cardiovascular comorbidities.11,13,16
Asciminib will certainly drive further efforts to optimize the treatment of CML. One area of interest to be investigated is the dual inhibition of BCR-ABL1 with asciminib and an ATP catalytic site TKI. Wylie et al. briefly describes the discovery of asciminib, and the original intention to use it along with another TKI in order to improve treatment outcomes and create a barrier to resistance.17 With preclinical analyses demonstrating an additive effect of asciminib in combination with imatinib, dasatinib, or nilotinib, Novartis continues to investigate the clinical feasibility of these combinations.17,18 There is also a case report for the use of combination asciminib plus bosutinib to restore control of resistant disease.19
Finally, the possibility exists for the development of mutations to the myristoyl pocket of BCR-ABL1, which can impact treatment success with asciminib.20 The occurrence of myristoyl pocket mutations in the phase 1 dose finding study were found to be lower than originally predicted from in vitro analyses, but should be considered in patients that do not show a response to or become refractory to asciminib. Myristoyl pocket mutation analysis will likely find its way into the clinical setting as the use of asciminib becomes better established in practice.
Conclusion
The introduction of asciminib into the armamentarium for CML provides another agent for individuals who have developed resistance to or are unable to tolerate previous TKI therapy. The pivotal trials for asciminib demonstrates a safe and effective option for patients with CP CML. Clinicians should be aware of the common AEs, particularly those requiring dose interruptions and/or adjustments. Future studies with asciminib as monotherapy or in combination with other TKIs will allow for a better understanding of asciminib’s place and utility in clinical practice.
REFERENCES
- Kantarjian H, O’Brien S, Jabbour E, et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: a single-institution historical experience. Blood. 2012; 119(9):1981-1987.
- Kantarjian HM, Baccarani M, Jabbour E, Saglio G, Cortes JE. Second-generation tyrosine kinase inhibitors: the future of frontline CML therapy. Clin Cancer Res. 2011; 17:1674-1683.
- Fava G, Morotti A, Dogliotti I, Saglio G, Rege-Cambrin G. Update on emerging treatments for chronic myeloid leukemia. Expert Opin. Emerging Drugs. 2015; 20:183-196.
- Miura M. Therapeutic drug monitoring of imatinib, nilotinib, and dasatinib for patients with chronic myeloid leukemia. Biol Pharm Bull. 2015; 38:645- 654.
- Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2016 updated on diagnosis, therapy and monitoring. Am J Hematol. 2016; 91:252-265.
- Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendatons for the management of chronic myeloid leukemia: 2013. Blood. 2013; 122:872-884.
- Schoepfer J, Jahnke W, Berellini G, et al. Discovery of asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J Med Chem. 2018; 61:8120-8135.
- US FDA. FDA approves asciminib for Philadelphia chromosome-positive chronic myeloid leukemia. October 29, 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-asciminib-philadelphia-chromosome-positive-chronic-myeloid-leukemia.
- O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009; 16:401-412.
- Scemblix (asciminib). Package Insert. Novartis Pharmaceuticals Corporation. 2021.
- Hughes TP, Mauro MJ, Cortes JE, et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N Engl J Med. 2019; 381:2315- 2326.
- Hoch M, Sato M, Zack J, et al. Pharmacokinetics of asciminib in individuals with hepatic or renal impairment. The Journal of Clinical Pharmacology. 2021; 61(11):1454-1465.
- Réa D, Mauro MJ, Boquimpani C, et al. A phase 3, open-label, randomized study of asciminib, a STAMP inhibitor, vs bosutinib in CML after 2 or more prior TKIs. Blood. 2021; 138(21):2031-2041.
- Mauro MJ, Minami Y, Réa D, et al. Efficacy and safety results from ASCEMBL, a multicenter, open-label, phase 3 study of asciminib, a first-in-class STAMP inhibitor, vs. bosutinib in patients with chronic myeloid leukemia in chronic phase after ≥2 prior tyrosine kinase inhibitors: updated after 48 weeks. Presented at ASH 2021; December 11-14, 2021. Abstract 310.
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology. Chronic Myeloid Leukemia Version 2.2022. November 15, 2021; National Comprehensive Cancer Network. Available from: https://www.nccn.org/professionals/physician_gls/pdf/cml.pdf.
- Cortes JE, Kim DW, Pinilla-Ibarz J, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. 2018; 132(4):393-404.
- Wylie AA, Schoepfer J, Jahnke W, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017; 543:733-737.
- A phase 1 study of oral ABL001 in patients with CML or Ph+ ALL. ClinicalTrials.gov identifier: NCT02081378. Updated November 30, 2021. https://clinicaltrials.gov/ct2/show/NCT02081378?term=02081378&draw=2&rank=1.
- Hall KH, Brooks A, Waller EK. Overcoming TKI resistance in a patient with chronic myeloid leukemia using combination BCR-ABL inhibition with asciminib and bosutinib. Am J Hematol. 2021; e293-e295.
- Lee BJ, Shah NP. Identification and characterization of activating ABL1 1b kinase mutations: impact on sensitivity to ATP-competitive and allosteric ABL1 inhibitors. Leukemia. 2017; 31:1096-1107.
From Residency to Career: What Should Your First “Job” Be?
Career Advice from Recent Residents
Cambree Fillis, PharmD, BCOP
Oncology Clinical Pharmacy Specialist
Molly Graveno, PharmD
Oncology Clinical Pharmacy Specialist
Grace Hsu, PharmD, BCOP
Oncology Clinical Pharmacy Specialist
Catherine Johnson, PharmD
Oncology Clinical Pharmacy Specialist
It is that exciting, yet daunting time of the year again. The search for your next professional endeavor is underway.
Is there an ideal first job for someone coming out of residency? What is the right type of job? Should I specialize or sub-specialize right out of the gates? Is it ok to jump into a management or academic position right away? To answers these questions, we received the advice of recent residents, a Post-Graduate Year 2 Oncology residency program director, and a hiring manager.
Identify Priorities
Similar to searching for the best-fit residency program, job hunting requires research and reflection of professional and personal priorities. It is important to identify what is most important to you. Start by asking yourself where and how you want to practice. Are you willing to step out of your comfort zone and move across the country for your future job? Do you prefer to practice solely inpatient or outpatient or would you rather have the opportunity to rotate? Is it important for you to practice as a sub-specialist among a large group of colleagues at an academic medical center or would you prefer to be the oncology pharmacist caring for patients with a variety of malignancies at a community hospital?
Also be sure to ask questions to others. Speak to individuals familiar with the institutions you are applying to and gain as much insight as you can on the position and the institutional culture. These tactics will aid you in identifying positions that will positively challenge you and promote your career growth through opportunities for teaching, mentorship, research, and leadership.
Get to Know Hiring Timelines
There is no set timeline or designated match day for positions after residency. This can be challenging, as some programs recruit in early winter and others wait until late spring. Knowing the different timelines will allow you to coordinate multiple interviews around the same time and prevent you from having an offer on the table requiring a decision to be made earlier than other upcoming interviews. Most importantly, do not worry if you do not have a position lined up by March. Oncology pharmacy positions are always being posted, and you will find something that aligns well with your aspirations.
Seek Mentorship
During the job search, use and build your network. Find two to three mentors who have taken similar steps in their career, know you as a person, and can give you their honest opinion. Discuss your priorities and timeline with them. Learn the tips and tricks they used to navigate the job market, and adopt the ones that work for you. Seek their feedback on your CV, letters of intent, and prepared presentation if they are required by the institutions you are applying to.
Overall, as you search for the position best for you, be mindful of your priorities, timelines, and available mentors- keep in mind that your first job does not have to be your last job. There will be plenty of time to find your dream job. You may even find that your definition of a ‘dream job’ changes with your priorities. Throughout the process, keep an open mind, stay organized, and apply to institutions where you can see yourself thrive.
Career Advice from a Post-Graduate Year 2 Residency Program Director
Wendy Ying Ci Hui, PharmD, BCOP
Clinical Oncology Pharmacy Specialist
PGY2 Oncology Pharmacy Residency Director
Kellogg Cancer Center, Northshore University Health System – Evanston Hospital
Evanston, IL
When it comes to the ideal first job out of residency, the answer is definitely different for everyone. The ideal job depends on your skills, interests and personal priorities.
Whether you are preparing to look for a job during your oncology residency or you have worked a few years post residency and are now looking for a change, it is important to conduct a self-assessment and examine your skills, interests and goals. This may bring back memories of the pre-residency questionnaires you have completed. Some of your responses may have changed, so it is good to reevaluate.
Some self-reflective questions to help guide you through the process may include the following:
| Professional skills, interest, and goals | Personal priorities |
|---|---|
| What are you passionate about? | What are your personal plans? |
| What brought you the most satisfaction? | Do current work responsibilities allow you to fulfill other life goals? |
| What do you like the least? | Is proximity to family and friends important to you? |
| What do you do well? | Do you have family obligations that may influence where to work or ability to travel? |
| What are your short and long-term career goals? | Where would you like to live? |
Rank your answers: which are must haves and which ones are negotiable?
Once you have a clear picture of your professional and personal goals, discuss with your trusted advisors: residency program director (RPD), preceptors, mentors, family, friends, etc. They will provide helpful insights as you review potential oncology pharmacist positions.
The next step is to start the search. You can look for opportunities at your current institution, through pharmacy organization websites (career pages), online recruitment services, institutional website job postings, pharmacy friends/mentors’ referrals, etc.
What if you are not offered the ideal position after residency? Should you settle for any job?
Urgency to accept a job offer depends on many factors. Do you have an interim job that will sustain you financially as you continue your search? If you are able to stay as a resource pharmacist in your current institution, then you may able to wait longer for the ideal job.
If you need a job right away, then weigh your offer with current needs and future considerations.
- Does the position offer the must haves?
- Is there potential to develop the position into your ideal job?
- Will this job help further your professional development?
- Are there more positives than negatives?
It is important to recognize there is no perfect job. An ideal position may still have features you are less enthusiastic about. Do not be too quick to conclude whether an opportunity is good for you or not If you are interested but unsure if you have the required qualifications or if the position meets your needs, then apply and see how things turn out. Most of the time, you will not know until you are actually working in that position. There is always something to learn from an experience, so keep an open mind. Your goals and priorities may also change with time. Therefore, the first job, even if less than ideal, will prepare you for the position that you will find fulfilling for the long run.
Career Advice from a Hiring Pharmacist
Jessica Unzaga, PharmD, BCPS, BCOP
Pharmacy Clinical Coordinator, Malignant Hematology & Stem Cell Transplant
Miami Cancer Institute, Baptist Health South Florida
Miami, FL
Graduates of a post-graduate year 2 (PGY2) residency program enter the workforce with a dynamic and well-rounded skill set in clinical oncology, administration, teaching, and research, to name a few. In the winter and spring months PGY2 residents are busy searching for their next steps, and are often looking for sub specialization in a specific area of interest aligned with their exposure as a resident or even in student rotations.
As a coordinator that contributes to hiring decisions at my institution, I feel that the most important quality in an applicant is the concept of job fit. Job fitness is determined by combining the prospective employee’s strengths, motivations, and experience and how they match with the needs of the position and work environment. Employees that fit well into the role, department, and institution are highly satisfied, productive, and contribute to a positive work culture.
PGY2 graduates come with a solid foundation of oncology pharmacy practice and when looking for their first role after residency it is important that they take the time to reflect on their strengths, opportunities, and goals for the next 5 years and how that matches their role of interest. Furthermore, oncology practice models differ across the country and different roles and practice settings call for different skill sets.
It is important that during the application and interview process the applicant seeks to fully understand the entity, department, and position for which they are applying. Is the position new to fill an unmet need or does it replace a pharmacist that was previously integrated in that role? What are the key deliverables? How will I be evaluated? These are some great starting points for an applicant to ask future employers when assessing job fitness.
Another significant factor for PGY2 graduates to consider is that pharmacy practice, even more so, specifically oncology practice, is rapidly evolving. We should expect that a decade from now oncology pharmacy practice may look a bit different than today, many institutions have established protocols and/or collaborative practice agreements, and in the future pharmacists may obtain nationwide provider status. Employers looking to hire new practitioners are seeking candidates that are internally motivated to continue learning, optimize patient care, and build and maintain strong interdisciplinary collaboration, in addition to transparency, flexibility, and resilience when faced with change. Establishing boundaries and work life balance is also key to maintaining positive mental and physical health. Mentors are also great resources that can offer valuable insight to help guide residents through the job search process.
Feature: Updates in Bladder Cancer
Lindsey Chippendale
PGY2 Hematology/Oncology Pharmacy Resident at Penn Medicine, University of Pennsylvania Health System
Philadelphia, Pennsylvania
Bladder cancer, or urothelial carcinoma, is the sixth most common cancer diagnosis in the United States with an estimated 83,730 new cases and 17,200 deaths in 2021. There is a 77.1% 5-year relative survival in patients diagnosed with urothelial carcinoma.1 Metastatic urothelial carcinoma has a 5-year overall survival rate of 18%.2 Transitional cell carcinoma is the most common bladder cancer histology, accounting for 90% of all bladder cancers. 1 About 5% of urothelial carcinomas are upper tract which are tumors that develop in the renal pelvis or in the ureter.3
Current Guideline Recommendations:
In muscle-invasive bladder cancer (MIBC) stages two and three, treatment consists of neoadjuvant cisplatin-based combination chemotherapy including gemcitabine and cisplatin or dose-dense methotrexate, vinblastine, doxorubicin, and cisplatin (DDMVAC) with radical cystectomy, cystectomy alone, or chemoradiotherapy, depending on the performance status of the patient and tolerability of chemotherapy.4
First-line therapy for metastatic disease is dependent upon if the patient is cisplatin eligible. If cisplatin-eligible, the preferred regimen would consist of the same cisplatin-based chemotherapy regimens used for neoadjuvant therapy. If cisplatin-ineligible, the recommended first-line therapy is platinum-based chemotherapy with gemcitabine and carboplatin. An alternative first-line option for cisplatin-ineligible patients is immunotherapy for patients with programmed death-ligand 1 (PD-L1) expression or patients who are not eligible for any platinum-containing chemotherapy.5
A significant number of metastatic patients are unable to tolerate cisplatin therapy due to comorbidities including renal dysfunction or poor performance status.6 This represents a need for less toxic therapies to improve survival rates. Here we will discuss emerging data for systemic therapy in urothelial carcinoma including chemotherapy, immunotherapy, targeted therapy, and antibody drug conjugates.
Adjuvant Chemotherapy for Upper Tract Urothelial Carcinoma (UTUC):
Platinum-based chemotherapy including gemcitabine in combination with either cisplatin or carboplatin can be considered for UTUC if neoadjuvant therapy was not given (National Comprehensive Cancer Network (NCCN) Category 2A).
Platinum-based chemotherapy is an option based on POUT, a phase III, randomized, parallel group trial that compared adjuvant platinum-based chemotherapy to surveillance in patients with pT2- 4, Nany or pTany, N1-3, non-metastatic UTUC after nephroureterectomy. Patients received four 21-day cycles of gemcitabine (1,000 mg/ m2) days 1 and 8 of each cycle in combination with cisplatin (70 mg/m2) or carboplatin (AUC 4.5 or 5) on day 1. The primary endpoint of disease-free survival was estimated to be 71% in the chemotherapy cohort and 46% with surveillance (P=0.001) at 3 years. Acute treatment-emergent adverse events > grade 3 were reported for 44% of patients who received chemotherapy versus 4% with surveillance. In the chemotherapy group, there was a greater incidence of > grade 3 decrease in neutrophils (36%), decrease in platelet count (10%), febrile neutropenia (6%), vomiting (6%), and nausea (6%).7
Nivolumab for Adjuvant Therapy:
Nivolumab is an anti-PD-1 monoclonal antibody that is FDA approved for adjuvant therapy following resection in urothelial carcinoma. In bladder cancer, the PD-1 receptor is expressed and is a potential therapeutic target. However, for this indication there is no required testing for PD-1 expression. Anti-PD-1 antibodies inhibit the PD-1 pathway and stimulate the immune system to produce antitumor effects.8 Nivolumab can be considered for use as an “other recommended regimen” (NCCN category 2A) for adjuvant systemic therapy in patients with high-risk pathology after cystectomy regardless if cisplatin-based neoadjuvant therapy was given. High risk was defined as pathological stage of pT3, pT4a, or pN+ and patient ineligible for or declined adjuvant cisplatin-based chemotherapy and who had not received neoadjuvant cisplatin-based chemotherapy, or pathological stage ypT2-ypT4a or ypN+ for patients who received neoadjuvant cisplatin.
Nivolumab is approved for this indication based on CheckMate 247, a phase III, randomized, parallel group clinical trial comparing nivolumab 240 mg intravenously (IV) versus placebo every two weeks for up to one year in patients with MIBC that had received radical surgery. Nivolumab was assessed in patients who had received neoadjuvant therapy as well as in those who had not received neoadjuvant therapy. The primary endpoint showed statistically significantly improved disease-free survival with nivolumab at 20.8 months compared to placebo at 10.8 months (P<0.001), regardless of PD-L1 expression. The most common adverse events were pruritus 23.1%, fatigue 17.4%, and diarrhea 16.8%.9
Erdafitinib
Erdafitinib is a tyrosine kinase inhibitor that inhibits fibroblast growth factor receptor (FRGR) 1 through 4 signaling and is FDA approved for locally advanced or metastatic urothelial carcinoma.10 FGFR mutations occur in 20% of the metastatic urothelial carcinoma patient population.11 Mutations in FGFR contribute to unregulated cell proliferation and oncogenesis. Erdafitinib is indicated for patients who have progressed during or after platinum-based chemotherapy and whose tumors have susceptible FGFR3 or FGFR2 genetic alterations. Erdafitinib is included in the NCCN guidelines as a category 2A recommendation.
Erdafitinib is approved based on the BLC2001 trial. This was an open-label, single arm, phase II study that assessed the use of erdafitinib in patients with locally advanced or metastatic urothelial carcinoma with at least one FGFR3 mutation or FGFR2/3 fusion.
Patients received erdafitinib 8 mg by mouth once daily with or without food. If serum phosphate levels were < 5.5 mg/dL and the patient was not experiencing any ocular adverse effects or any grade 2 or greater adverse reactions on day 14, the patient increased to 9 mg once daily due to phase I study results showing an improved response rate with this dose. The primary endpoint of confirmed response rate occurred in 40% of patients. Median progression-free survival was 5.5 months, and median overall survival was 13.8 months.12 Adverse events of interest and management are detailed below.
Adverse effects (Package Insert %)
- Hyperphosphatemia (76%): Restrict phosphate intake to 600-800 mg daily. Monitor phosphate levels monthly. If serum phosphate > 7 mg/dL, consider adding an oral phosphate binder until phosphate returns to < 5.5 mg/dL.
- Ocular disorders (25%): Advise patients to either use artificial tears or hydrating or lubricating eye gels or ointments every 2 hours while awake to prevent dry eyes. Hold treatment if central serous retinopathy occurs. Permanently discontinue if it does not resolve in 4 weeks or if grade 4. Monitor with monthly ophthalmological examinations during the first 4 months of therapy and every 3 months after.
Clinical Pearl
- Erdafitinib inhibits FGFR signaling in the renal proximal tubule where it impairs the function of the sodium dependent phosphate co-transporter, inhibiting phosphate reabsorption and leading to hyperphosphatemia.
Enfortumab Vedotin
Enfortumab vedotin (EV) is an antibody drug conjugate (ADC) that is FDA approved for locally advanced or metastatic urothelial carcinoma. EV targets nectin-4, which is a cell-adhesion protein that is located on cell surfaces and is highly expressed in urothelial carcinoma. The human IgG1 antibody enfortumab binds to nectin-4 and is internalized, where monomethyl auristatin E (MMAE), a microtubule-disrupting agent, is then released and induces cell cycle arrest and cell death.13 EV is preferred for the treatment of adult patients who are ineligible for cisplatin-containing chemotherapy and have previously received one or more prior lines of therapy (NCCN category 2A for second line therapy, category 1 for subsequent therapy).
The NCCN recommendation for EV is based on the EV-301 open-label, randomized, phase III, parallel group clinical trial comparing EV versus chemotherapy in patients with locally advanced or metastatic urothelial carcinoma who had previously received platinum-based chemotherapy and had disease progression during or after treatment with a PD-1 or PD-L1 inhibitor. Enfortumab vedotin was administered at 1.25 mg/kg (max of 125 mg for patients >100 kg) of body weight IV infusion over 30 minutes on days 1, 8, and 15 of a 28-day cycle until disease progression or unacceptable toxicity. The chemotherapy cohort received one of the following options: docetaxel, paclitaxel, or vinflunine. EV was statistically significant in the primary and secondary endpoints in improving overall survival with a median of 12.88 months versus 8.97 months (P=0.001) and progression-free survival with a median of 5.55 months versus 3.71 months (P<0.001), respectively. Adverse events in the EV cohort included maculopapular rash, peripheral neuropathy, and hyperglycemia.14 Adverse events of interest and management are detailed below.
Adverse effects (Package Insert %)
- Hyperglycemia (16%): If blood glucose is >250 mg/dL hold therapy. Grade 3 or 4 hyperglycemia occurred in about 8% of patients, and incidence increased in patients with higher body mass index and higher baseline A1C.
- Peripheral neuropathy (49%): Consider dose reduction or holding therapy if peripheral neuropathy occurs. Discontinue therapy in patients with grade 3 or greater peripheral neuropathy.
- Ocular disorders (46%): Ocular adverse events include keratitis, blurred vision, and limbal stem cell deficiency. Consider artificial tears for dry eye prophylaxis. Consider ophthalmologic evaluation, dose reduction, or holding therapy if ocular symptoms occur.
- Skin reactions (54%): Grade 3 to 4 skin reactions occurred in 10% of patients. Consider topical steroids and antihistamines for treatment. Hold therapy for grade 3 skin reactions and permanently discontinue therapy in patients that have grade 4 or recurrent grade 3 skin reactions.
- Infusion site extravasation (1.3%): Ensure adequate venous access prior to infusing medication. Monitor for extravasation during administration. Reactions can be delayed, with erythema, swelling, and pain worsening 2 to 7 days after extravasation.
Clinical Pearls
- Though there is a high incidence of peripheral neuropathy with EV, it is largely reversible. Phase I and II studies had 76% of patients with a peripheral neuropathy adverse reaction return to baseline or grade 1 at last follow up.13
- In clinical trials, diabetic ketoacidosis (DKA) occurred in patients with and without preexisting diabetes mellitus. Monitor for hyperglycemia and DKA in all patients.
- Nectin-4 is expressed in the skin, attributing to the skin toxicity effects seen with use of EV.
Sacituzumab Govitecan
Sacituzumab govitecan is an ADC that is FDA approved for locally advanced or metastatic urothelial carcinoma. Sacituzumab govitecan is directed against trophoblast cell surface antigen 2 (Trop- 2). Trop-2 is a transmembrane glycoprotein that is expressed on epithelial cancer cells and stimulates cell proliferation. Sacituzumab govitecan binds to Trop-2 and is internalized, releasing SN-38, a topoisomerase 1 inhibitor, leading to DNA damage and cell death.16 Sacituzumab govitecan is indicated for use as subsequent therapy as an “other recommended regimen” (NCCN category 2A) after treatment with a platinum-based therapy and a checkpoint inhibitor if enfortumab vedotin or erdafitinib are not appropriate for the patient.
Sacituzumab govitecan is approved based on TROPHY-U-01, an open-label, single arm, phase II trial that assessed sacituzumab govitecan in patients with locally advanced, unresectable, or metastatic urothelial carcinoma with disease progression following a platinum-based regimen and checkpoint inhibitor therapy.
Patients received sacituzumab govitecan 10 mg/kg administered as an IV infusion once weekly on days 1 and 8 of a 21-day cycle, and continued treatment until disease progression or unacceptable toxicity. The primary endpoint of overall response rate was 27% with responses lasting for a median of 7.2 months. Secondary endpoints were median progression-free survival (5.4 months), and median overall survival (10.9 months).17 Adverse events of interest and management are detailed below.
Adverse effects (Package Insert %)
- Neutropenia (61%): Hold therapy for absolute neutrophil count below 1500/mm3 on day 1 of any cycle, neutrophil count below 1000/mm3 on day 8 of any cycle, or for neutropenic fever. Consider dose requirements for neutropenia.
- Diarrhea (65%): If negative for infectious causes of diarrhea, initiate loperamide 4 mg followed by 2 mg with every episode of diarrhea (maximum 16 mg daily). Consider supportive measures for fluid and electrolyte substitution. If patient has cholinergic response to treatment can provide atropine premedication for subsequent treatments. Hold therapy for grade 3 to 4 diarrhea at time of scheduled treatment and resume when resolved to grade < 1.
- Hypersensitivity and infusion-related reactions (37%): Reactions typically occur within 24 hours of dose administration. Pre-medicate and have emergency medications to treat infusion-related reactions including anaphylaxis available for immediate use. Permanently discontinue therapy for grade 4 infusion-related reactions.
- Nausea and vomiting (66%): Hold therapy for grade 3 nausea or grade 3 to 4 vomiting at time of scheduled administration and resume when resolved to grade <1. Ensure patients are provided take-home medications for prevention and treatment of nausea and vomiting.
Clinical Pearls
- Administer the first infusion over 3 hours. Monitor patient throughout infusion and at least 30 minutes after each dose for infusion-related reactions. For subsequent infusions, administer over 1 to 2 hours if prior infusions were tolerated.
- Pre-medicate with a two or three drug antiemetic combination to prevent nausea and vomiting and use antipyretics and H1/ H2 antagonists to prevent an infusion related reaction. If a prior infusion-related reaction occurred, use corticosteroids (hydrocortisone 50 mg or equivalent by mouth or IV) for subsequent infusions.
- SN-38 is an irinotecan metabolite, therefore the adverse event profile of sacituzumab govitecan is similar to irinotecan, including neutropenia and diarrhea.
- Patients who are homozygous for uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at an increased risk for adverse events including febrile neutropenia, neutropenia, and anemia. In patients with unexpected severe or acute early-onset adverse reactions, this may indicate reduced UGT1A1 enzyme activity. Recommendations are to hold therapy or permanently discontinue based on clinical assessment of observed reactions.
Conclusion
Within the past few years the treatment options for metastatic urothelial carcinoma have expanded greatly. Overall survival rates have extended from 7-10 months to 12-13 months and response rates have doubled from 20% to 40% with these new therapies. FGFR targeted therapy and antibody drug conjugates provide alternative options with different mechanisms of action to utilize in patients with chemotherapy resistance.
Ongoing trials are assessing use of immunotherapy in combination with targeted therapy or chemotherapy. Future treatment strategies may also include incorporation of additional targeted agents including poly ADP-ribose polymerase enzyme inhibitor (PARP) inhibitors, vascular endothelial growth factor (VEGF) inhibitors, and mammalian target of rapamycin (mTOR) inhibitors, and lean more towards focusing on a personalized targeted approach to treatment in patients with metastatic urothelial carcinoma.2
REFERENCES
- SEER Stat Fact Sheets: Bladder Cancer. 2021. https://seer.cancer.gov/statfacts/html/urinb.html.
- Mollica V, Rizzo A, Montironi R, et al. Current strategies and novel therapeutic approaches for metastatic urothelial carcinoma. Cancers (Basel). 2020;12(6):1449.
- Collà Ruvolo C, Nocera L, Stolzenbach LF, et al. Incidence and survival rates of contemporary patients with invasive upper tract urothelial carcinoma. Eur Urol Oncol. 2021;4(5):792-801.
- Von der Maase H, Hansen SW, Roberts JT, et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol. 2000;18(17):3068-77.
- NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Bladder Cancer. V.6.2021, 12/06/21, 2020 National Comprehensive Cancer Network, Inc. https://www.nccn.org/professionals/physician_gls/pdf/bladder.pdf.
- Galsky MD, Hahn NM, Rosenberg J, et al. Treatment of patients with metastatic urothelial cancer “unfit” for cisplatin-based chemotherapy. J Clin Onco.l 2011;29:2432–38.
- Birtle A, Johnson M, Chester J, et al. Adjuvant chemotherapy in upper tract urothelial carcinoma (the POUT trial): a phase 3, open-label, randomized controlled trial. Lancet. 2020;395(10232):1268-77.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12:252–64.
- Bajorin DF, Witjes JA, Gschwend JE, et al. Adjuvant nivolumab versus placebo in muscle-invasive urothelial carcinoma. N Engl J Med. 2021;384(22):2102-14.
- Balversa (erdafitinib) [prescribing information]. Horsham, PA: Janssen Products, LP; July 2020.
- Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15:25-41
- Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381(4):338-48.
- Padcev (enfortumab vedotin) [prescribing information]. Northbrook, IL: Astellas Pharma US Inc; July 2021.
- Powles T, Rosenberg JE, Sonpavde GP, et al. Enfortumab vedotin in previously treated advanced urothelial carcinoma. N Engl J Med. 2021;384(12):1125-35.
- Rosenberg JE, O’Donnell PH, Balar AV, et al. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J Clin Oncol.2019;37:2592-2600.
- Trodelvy (sacituzumab govitecan) [prescribing information]. Morris Plains, NJ: Gilead Sciences Inc; October 2021.
- Tagawa ST, Balar AV, Petrylak DP, et al. TROPHY-U-01: A phase II open-label study of sacituzumab govitecan in patients with metastatic urothelial carcinoma progressing after platinum-based chemotherapy and checkpoint inhibitors. J Clin Oncol. 2021;39(22):2474-85.
Connection, Community, and Care Navigation Through the Eyes of the Leukemia & Lymphoma Society
Laura Cannon, PharmD, MPH, BCOP
Clinical Assistant Professor and Oncology Pharmacist
The University of Texas at Austin College of Pharmacy and Dell Medical School Livestrong Cancer Institutes
Austin, TX
Marin Abousaud, PharmD, BCOP
Oncology Clinical Pharmacy Specialist, Head and Neck Cancer
Emory Healthcare, Winship Cancer Institute
Atlanta, GA
As humans, we are built for connection. We need it to provide support, to foster growth, and to share life’s ups and downs. The needs of a cancer patient are no different. Often, the difficulty lies in finding someone that understands your experience, can relate to your concerns, and can provide validation. It is not always possible to find someone of similar age or diagnosis within your own hospital or even within your own community. The Leukemia & Lymphoma Society (LLS) is just one of many patient advocacy organizations that have identified this need and provided online resources to create necessary connection and support for patients and their loved ones. The LLS mission is to cure leukemia, lymphoma, Hodgkin’s disease and myeloma, and improve the quality of life of patients and their families.
LLS Community is an online platform where more than 16,000 patients, caregivers, and healthcare providers currently subscribe as members. This community is a virtual space where members can interact with each other in numerous ways including posting questions, links, and successes on a community news feed or connecting with an Information Specialist such as a nurse navigator or social worker to assist with care navigation. By becoming a member of LLS Community, you have access to numerous educational resources and first-hand experience from patients, caregivers, and healthcare providers across the country at your fingertips.
In addition to the large LLS Community, members have the opportunity to join smaller groups related to their specific diagnosis or demographics such as BIPOC, LGBTQI, Caregivers, Parents, Veterans, or those interested in Fertility and Pregnancy. Allowing members to connect with a community can be more valuable than any educational resource and can be a source of inspiration and hope for those navigating the cancer journey. Tricia Hernandez, who currently serves at the Senior Manager of Community Engagement for LLS, says, “I feel like patients who are involved in the community have a greater opportunity to connect. Almost all patients join and say, ‘I need to meet someone else who is going through what I’m going through’.” LLS Community allows patients to do just that, which can be so valuable, especially during times where patients may feel anxious, alone, or isolated.
LLS has employed many strategies to foster engagement on their online community platform such as posing a ‘Question of the Day’ to promote interaction and discussion. They also focus on sharing timely articles to the news feed that patients may find relevant. For example, sharing articles from a national meeting while it’s occurring or articles related to the effect of the COVID-19 pandemic on cancer patients. Any member is able to comment directly on these posts with additional findings or questions, allowing members to actively participate in becoming more knowledgeable about topics that impact their care. Posts may also serve to encourage discussion amongst patients and healthcare providers at the local level by exposing patients to timely resources.
In addition to LLS Community, the organization also provides virtual education programs including a national blood cancer conference, national webinars and local education sessions. A recent change has allowed content to be available online for virtual blood cancer conferences even after the initial conference took place, which increased the accessibility of educational resources for those who registered.
As pharmacists, we have all experienced the hunger patients and caregivers have for educational information and for someone to relate to their experiences. LLS Community offers direct access to these resources that other patients, caregivers, and healthcare providers have found to be helpful. Another resource that is often lacking is a common space to track information related to a patient’s cancer diagnosis. Through the use of the LLS Health Manager™ mobile application, patients have the ability to track side effects, medications, questions, and food and water intake. The application will generate a report based on your tracking information and even provides the option to share this information with your caregiver and healthcare team. In addition to LLS Community, this application allows patients and caregivers to take an active role in their cancer journey.
We all know navigating a cancer diagnosis can be overwhelming for all parties involved. These examples, through the lens of LLS, provide an overview of some of the many resources available through patient advocacy organizations. The more we can familiarize ourselves with available resources, the more we can continue to work together to provide the best support to our patients and their loved ones. To become a member of LLS Community please visit communityview.LLS.org. The LLS Health Manager™ application is available as a free download through your smart phone application store.
Tricia Hernandez, MS has worked with The Leukemia & Lymphoma Society for the past six years in roles as a Patient & Community Outreach Manager and now Senior Manager of Community Engagement. In these roles, she works directly with patients, caregivers and healthcare professionals to share support and education programming and resources and increase access to care for underserved populations. She manages the online LLS Community, which provides a gathering space to share hope, support and information. Tricia is also an 18-year lymphoma survivor.
Prospective Evaluation of High-Dose Methotrexate Pharmacokinetics in Adult Patients with Lymphoma Using Novel Determinants of Kidney Function
Jason N. Barreto, PharmD, MSc, BCPS
Assistant Professor of Medicine and Pharmacy,
Mayo Clinic School of Medicine
Clinical Pharmacist - Hematology and Oncology
Mayo Clinic
Rochester, MN
Introduction
High-dose methotrexate (≥1 gram/meter2 [g/m2]; HDMTX) is a cornerstone of treatment for central nervous system (CNS) lymphoma.1 It has also been integrated into the management of systemic lymphoma as prophylaxis for patients who are at high risk of disease relapse in the CNS.2 Despite more than 60 years of utilization, the optimal dose, administration, monitoring, and supportive measures have yet to be determined.3
Methotrexate (MTX) dose selection is a delicate balance between optimizing tumor kill without subjecting the host to excess toxicity.4 The standard of care for MTX dose derivation utilizes serum creatinine as a surrogate for glomerular filtration rate (GFR).5 As the terminal byproduct of skeletal muscle metabolism, non-renal determinants, including altered muscle mass, deconditioning, and malnutrition, can decrease the accuracy of serum creatinine-based GFR estimation in patients with cancer.6-8
Cystatin C is a serum marker of GFR that is less dependent on age, sex, race, or muscle mass than creatinine.9 The use of cystatin C to inform drug dosing and monitor dynamic kidney function has recently increased and cystatin C has successfully been used in conjunction with, or as an alternative to, creatinine to dose antineoplastics, including carboplatin and topotecan.10-13 Given that there is also data to support using cystatin C to estimate GFR prior to HDMTX delivery in patients with creatinine-based estimated that seem suspicious14, we sought to understand the relationship between HDMTX pharmacokinetics and cystatin C-inclusive GFR estimation equations.
Methods
Our prospective, single-center study included adult patients (≥18 years old) with histologically confirmed lymphoma admitted for administration of HDMTX. Patients prescribed a HDMTX infusion administered longer than 4 hours, patients admitted with new acute kidney injury before the HDMTX infusion, or those receiving renal replacement therapy were excluded.
After enrollment, we collected biospecimen samples at fixed time points from before HDMTX delivery until either 96-hours after the infusion or patient discharge from the hospital. The study procedures did not affect the routine clinical care for patients. Research-related laboratory results were suppressed from the electronic health record and were available only to study personnel. The interdisciplinary care team selected the HDMTX-inclusive treatment without regard to the biomarker levels. Per hospital protocol, serum MTX concentration monitoring began at 48-hours after the drug infusion and then occurred once daily until <0.1 μmol/L, indicative of discharge from the hospital, barring other clinical circumstances. Patients were followed until the next HDMTX dose or for 30 days, whichever occurred first.
The pharmacokinetics of MTX were estimated by standard non-compartmental analysis using the program Phoenix® WinNonlin® Version 8.1(Certara Corporation, Princeton, NJ). The area under the concentration-time curve (AUC) was calculated using the linear trapezoidal approximation. MTX plasma clearance (CLp) was calculated as Dose/AUC. The Spearman correlation coefficient was used to compare pharmacokinetic parameters and patient characteristics. We fit linear regression models to determine whether varying the approach to baseline kidney assessment (eGFR based on Cockcroft Gault (C-G) or Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equations using serum creatinine, cystatin C, or both biomarkers) improved the prediction of drug clearance.
Data were summarized using mean ± standard deviation (SD) or median with the interquartile range (IQR) depending on the distribution. Frequencies (percentages) were used to describe discrete data. The Wilcoxon signed-rank test was used to detect intra-individual differences in kidney function estimates.
Results and discussion
A total of 80 individuals met eligibility criteria and were enrolled. Patients had a median age of 69 (IQR: 59-76) years, 54 (68%) were male, and 74 (93%) were white. The median body weight of the patient population was 80.5 kg (IQR: 70-92) and the median BSA was 1.97 (IQR: 1.80-2.14) as calculated by the Du Bois Method. There were five patients (6%) with a baseline diagnosis of CKD. Baseline estimated kidney function differed according to equation utilized and ranged between a mean of 83 mL/min when calculated using CKD-EPI eGFR-CysC and 99 mL/min when calculated with C-G eGFR.
Seven patients had extremely high values for MTX clearance which influenced these relationships [median clearance 18.8 (IQR: 13.2-38.1) L/hr in these seven patients versus 4.8 (IQR: 3.8-5.8) L/hr in the remaining 73 patients]. Analytical techniques were reviewed and a detailed chart review was performed. Neither revealed common features, nor explanations, for the extreme drug clearance values in these patients. A sensitivity analysis was performed after considering these seven patients to be outliers and excluding them, which strengthened the observed correlations between MTX clearance and kidney function estimates. See Table 1 below.
In the full cohort of 80 patients, there was a modest relationship between MTX clearance and baseline estimated kidney function when calculated using creatinine, cystatin C, or both biomarkers. Interestingly, we found that the eGFR based on cystatin C, whether expressed in mL/min or mL/ min/1.73m2, predicted MTX clearance better than creatinine-based estimating equations (Table 1).
Additionally, the eGFR equation utilizing both serum creatinine and cystatin C also showed a stronger correlation with MTX clearance than the Cockcroft Gault estimated creatinine clearance. This effect seems primarily driven by the cystatin C component, given that the correlation improved when comparing the eGFR based on serum creatinine and the eGFR incorporating both serum creatinine and cystatin C. This differs considerably from the current approach to MTX dosing in clinical practice that relies solely on serum creatinine and estimated creatinine clearance based on the Cockcroft-Gault equation despite pharmacokinetic studies describing the poor performance of serum creatinine and creatinine clearance as a marker of MTX elimination.
Conclusion
This prospective, single-center pharmacokinetic clinical trial demonstrated that novel eGFR equations involving cystatin C appeared to more strongly correlate with MTX clearance than eGFR equations based on serum creatinine alone, highlighting a potential opportunity for enhancing the precision of MTX dosing using novel renal biomarkers. Future research should confirm these findings through population pharmacokinetic modeling analysis and observation of clinical outcomes after doses of HDMTX that utilize creatinine-based kidney function estimation compared to HDMTX doses calculated with cystatin-c-inclusive kidney function estimating equations.
Table 1. Correlation and 95% confidence intervals for glomerular filtrate rate estimation equations with methotrexate clearance.
| Entire population (n = 80) | Population after excluding outliers (n = 73) | |||
|---|---|---|---|---|
| MTX clearance in L/hr Correlation (95% CI) | MTX clearance in L/hr/BSA Correlation (95% CI) | MTX clearance in L/hr Correlation (95% CI) | MTX clearance in L/hr/BSA Correlation (95% CI) | |
| eCrClCG (mL/min) | 0.11 (-0.11, 0.33) | 0.13 (-0.09, 0.34) | 0.33 (0.10, 0.52) | 0.36 (0.14, 0.55) |
| eGFRcr (mL/min) | 0.17 (-0.05, 0.38) | 0.19 (-0.04, 0.39) | 0.38 (0.17, 0.56) | 0.39 (0.17, 0.57) |
| eGFRcys (mL/min) | 0.30 (0.09, 0.49) | 0.31 (0.09, 0.49) | 0.52 (0.33, 0.67) | 0.48 (0.28, 0.64) |
| eGFRcr-cys (mL/min) | 0.28 (0.06, 0.47) | 0.28 (0.07, 0.47) | 0.51 (0.31, 0.66) | 0.48 (0.29, 0.64) |
| eGFRcr (mL/min/1.73 m2) | 0.14 (-0.08, 0.35) | 0.15 (-0.07, 0.36) | 0.24 (0.01, 0.45) | 0.27 (0.04, 0.47) |
| eGFRcys (mL/min/1.73 m2) | 0.27 (0.05, 0.46) | 0.27 (0.06, 0.46) | 0.39 (0.18, 0.57) | 0.37 (0.16, 0.56) |
| eGFRcr-cys (mL/min/1.73 m2) | 0.24 (0.02, 0.44) | 0.25 (0.03, 0.44) | 0.36 (0.15, 0.55) | 0.36 (0.15, 0.55) |
| Abbreviations: BSA, body surface area; CI, confidence interval; MTX, methotrexate; eCrClCG, estimated creatinine clearance based on the Cockcroft-Gault formula; eGFR, estimated glomerular filtration rate based on the Chronic Kidney Disease Epidemiology Collaborative equation utilizing cr, serum creatinine, cys, cystatin C, or cr-cys, both serum creatinine and cystatin C; L/h, liters per hour, L/h/BSA, liters per hour per BSA; mL/min, milliliters per minute; mL/min/1.73 m2, milliliters per minute per 1.73 m2; MTX, methotrexate. | ||||
REFERENCES
- Rubenstein JL, Gupta NK, Mannis GN, et al: How I treat CNS lymphomas. Blood 122:2318-30, 2013
- Abramson JS, Hellmann M, Barnes JA, et al: Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 116:4283-90, 2010
- Howard SC, McCormick J, Pui CH, et al: Preventing and Managing Toxicities of High-Dose Methotrexate. Oncologist 21:1471-1482, 2016
- Warren JB: Translating the dose response into risk and benefit. Br J Clin Pharmacol 85:2187-2193, 2019
- Batchelor T, Carson K, O’Neill A, et al: Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96-07. J Clin Oncol 21:1044-9, 2003
- Lameire N, Van Biesen W, Vanholder R: Acute renal problems in the critically ill cancer patient. Curr Opin Crit Care 14:635-46, 2008
- Levey AS, Inker LA: Assessment of Glomerular Filtration Rate in Health and Disease: A State of the Art Review. Clin Pharmacol Ther 102:405-419, 2017
- Kashani K, Rosner MH, Ostermann M: Creatinine: From physiology to clinical application. Eur J Intern Med 72:9-14, 2020
- Teaford HR, Barreto JN, Vollmer KJ, et al: Cystatin C: A Primer for Pharmacists. Pharmacy (Basel) 8, 2020
- Teaford HR, Rule AD, Mara KC, et al: Patterns of Cystatin C Uptake and Use Across and Within Hospitals. Mayo Clin Proc 95:1649-1659, 2020
- Bretagne M, Jouinot A, Durand JP, et al: Estimation of glomerular filtration rate in cancer patients with abnormal body composition and relation with carboplatin toxicity. Cancer Chemother Pharmacol 80:45-53, 2017
- White-Koning M, Paludetto MN, Le Louedec F, et al: Formulae recently proposed to estimate renal glomerular filtration rate improve the prediction of carboplatin clearance. Cancer Chemother Pharmacol 85:585- 592, 2020
- Hoppe A, Seronie-Vivien S, Thomas F, et al: Serum cystatin C is a better marker of topotecan clearance than serum creatinine. Clin Cancer Res 11:3038-44, 2005
- Barreto JN, McClanahan AL, Rule AD, et al: Incorporating Cystatin C to Predict Methotrexate Elimination in Patients with CNS Lymphoma and Suspicious Renal Function. Case Rep Hematol 2018:7169897, 2018
The Butterfly Effect: Escalating Adjuvant Endocrine Therapy with monarchE
Allison Butts, PharmD, BCOP
Clinical Coordinator, Oncology Pharmacy; Director,
PGY2 Oncology Residency Program,
UK HealthCare- Markey Cancer Center,
Lexington, KY
Hormone receptor positive (HR+), human epidermal growth factor receptor 2 negative (HER2-) breast cancer represents nearly 70% of breast cancer cases, the vast majority of which are diagnosed in early stages. The 5-year relative survival for this cohort of patients exceeds 94% when all stages are considered together. However, this figure drops from 100% for localized disease at presentation to 89.9% with regional disease, and further to 30.6% for metastatic disease.1 In addition to surgery +/- radiation, treatment of early-stage HR+ breast cancer varies by stage and may include chemotherapy plus endocrine therapy, or endocrine therapy alone (aromatase inhibitors or tamoxifen, +/- ovarian suppression). Adjuvant bisphosphonate therapy may also be added to reduce the risk of bone recurrence for high-risk patients.2
Meaningful data have been published to guide treatment selection based on risk of recurrence for early-stage HR+ breast cancer patients. The TAILORx trial validated the use of the 21-gene breast cancer assay, Oncotype DX®, to predict chemotherapy benefit based on recurrence risk score in HR+, lymph node negative patients.3 RxPONDER followed, which demonstrated a similar ability to predict chemotherapy benefit using the Oncotype recurrence score in patients with one to three positive lymph nodes.4 The MINDACT trial provided data on the use of the 70-gene signature test, MammaPrint®, as a tool to tailor treatment plans for patients with early-stage HR+ with up to three positive lymph nodes.5 Together, these clinical trials have reduced the amount of chemotherapy that is administered to patients with low-risk HR+ breast cancer, while maintaining favorable disease recurrence and survival outcomes.
Unfortunately, about 20% of patients treated with adjuvant endocrine therapy will develop a breast cancer recurrence within 10 years.6 As a result, much work is being done to identify approaches to enhance or escalate treatment of high-risk HR+ breast cancer. Addition of a CDK4/6 inhibitor to adjuvant endocrine therapy has been investigated in three key trials: PALLAS, PENELOPE-B, and monarchE.
CDK4/6 inhibitors are a mainstay in the treatment of metastatic HR+ breast cancer.2 Upon binding of estrogen to the estrogen receptor, cyclin D1 expression is increased, along with activation of cyclin-dependent kinases, ultimately resulting in phosphorylation of the retinoblastoma protein (pRb).7 CDK4 and CDK6, in complex with cyclin D, drive cell progression from the G1 to S phase. Therefore, CKD4 and CDK6 are attractive drug targets due to the ability to arrest the cell cycle and block cell growth.8
The phase 3 PALLAS trial randomized patients with stage 2 or 3 HR+ breast cancer (n=5760) to receive palbociclib for 2 years in addition to 5-10 years of endocrine therapy, or endocrine therapy alone for 5-10 years. At the time of the final analysis, 4-year invasive disease-free survival (IDFS) was 84.2% in the palbociclib group and 84.5% in the control group (p=0.65).9 The phase 3 PENELOPE-B trial enrolled high-risk HR+ patients with residual disease after neoadjuvant chemotherapy (n=1250). Patients were treated with palbociclib for 1 year in addition to 5-10 years of endocrine therapy, or endocrine therapy alone. No difference was seen between the groups at 4 years for IDFS (73% vs. 72.4%, p=0.53).10
Recently, the phase 3 monarchE trial randomized high-risk HR+ patients (n=5637) to 2 years of abemaciclib in addition to 5-10 years of endocrine therapy, or endocrine therapy alone. To be included, patients had to have at least four positive lymph nodes or one to three positive nodes with one of the following additional risk factors: tumor size ≥5 cm or grade 3 histology (Cohort 1), or one to three positive nodes and Ki-67 ≥ 20% (Cohort 2). Patients may have received up to 12 weeks of endocrine therapy prior to randomization. Radiation, adjuvant, and neoadjuvant chemotherapy was received in 95.4%, 58.3%, and 37.0% of the study population, respectively. Patients were of a median age of 51.0 years and the majority were post-menopausal at diagnosis (56.5%). Aromatase inhibitors were the most commonly prescribed endocrine therapy (68.3%, including 14.2% treated concurrently with ovarian suppression).11
Unlike PALLAS and PENELOPE-B, monarchE reported a benefit in invasive disease-free survival (IDFS). At 2 years, addition of abemaciclib improved IDFS from 89.3% to 92.3% (p=0.0009) in the intent-to-treat population. Furthermore, distant recurrence-free survival (DRFS) was increased from 90.8% to 93.8% (p=0.0009).12 This benefit was maintained at 3 years for both IDFS (88.8% vs. 83.4%, p<0.0001) and DRFS (86.1% vs 90.3%, p<0.0001). When patients in Cohort 1 were stratified based on Ki-67 index, those who were Ki-67-high had an absolute benefit in 3-year IDFS of 7.1% (HR 0.63, 91% CI 0.49-0.80). In contrast, patients in Cohort 1 with a low Ki-67 index demonstrated a less pronounced absolute benefit of 4.5% (HR 0.70, 91% CI 0.51-0.98). While both subsets of patients derived a benefit from abemaciclib, the magnitude of benefit was reduced based on this biomarker.13 Based on the results of the monarchE trial, the FDA approved abemaciclib, in combination with endocrine therapy, for the adjuvant treatment of adult patients with HR+, node-positive, early breast cancer at high risk of recurrence and a Ki-67 score ≥20% on October 12, 2021. The Ki-67 IHC MIB-1 pharmDx assay was approved as a companion diagnostic.14 Notably, the FDA-approved indication may expand pending additional follow-up data such as overall survival in the intent-to-treat population.
There are several potential explanations for the discordant results between these three trials. First, the definition of “high-risk” was variable between trials and may have skewed the efficacy results. The duration of adjuvant CDK4/6 inhibition notably varied from 2 years in PALLAS and monarchE to 1 year in Penelope-B. The phase 3 NATALEE trial is ongoing, which will explore the activity of 3 years of ribociclib in the adjuvant setting, possibly providing more clarity on this issue.15 Next, the discontinuation rates varied between trials, with 27.7% of patients discontinuing abemaciclib early in monarchE, and 42% and 20% discontinuing palbociclib in PALLAS and PENELOPE-B, respectively.9,10,12 Lastly, the duration of follow-up differed between studies. Importantly, the improvement in IDFS observed in PENELOPE-B declined over time, with a 4.3% improvement at 2 years, 3.5% at 3 years, and 0.6% 4 years.10 The authors of monarchE have demonstrated sustained results from 2 to 3 years but longer follow-up is needed for each of these landmark trials.
The landscape of treatment options for HR+ breast cancer is continually evolving as we learn more about opportunities to tailor treatments based on risk stratification. De-escalation of therapy in low-risk early-stage disease using genomic assays is now commonplace in clinical practice. Identifying high-risk patients for escalation of therapy is now a major focus area for clinical research. The monarchE data supports incorporating 2 years of adjuvant abemaciclib therapy in select high-risk patients, making abemaciclib the first drug to receive FDA-approval for adjuvant hormone-receptor positive breast cancer in over 15 years.14 However, conflicting results from other large studies should not be ignored and require critical evaluation.
REFERENCES
- Cancer Stat Facts: Female Breast Cancer Subtypes. https://seer.cancer.gov/statfacts/html/breast-subtypes.html. Accessed 3 December 2021.
- NCCN Clinical Practice Guidelines in Oncology. Breast Cancer. Version 1.2022. https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed 3 December 2021.
- Sparano JA, et al. N Engl J Med. 2018;379:111-121.
- Kalinsky K, et al. San Antonio Breast Cancer Symposium. 2020;Abstract GS3-00.
- Cardosa F, et al. N Engl J Med. 2016;375:717-29.
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Lancet. 2015;386:1341-52.
- Foster JS, et al. Mol Cell Biol. 2001;21:794-810.
- Sherr CJ, Beach D, Shapiro GI. Cancer Discov. 2016;6:353-367.
- Gnant M, et al. San Antonio Breast Cancer Symposium. 2021;Abstract GS1-07.
- Loibl S, et al. J Clin Oncol. 2021;39:1518-1530.
- Johnston SR, et al. J Clin Oncol. 2021;38:3987-98.
- O’Shaughnessy JA, et al. San Antonio Breast Cancer Symposium. 2020;Abstract GS1-01.
- Harbeck N, et al. Ann Oncol. 2021;32:1571-81.
- US Food and Drug Administration. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-abemaciclib-endocrine-therapy-early-breast-cancer. Accessed 3 December 2021.
- ClinicalTrials.gov. Accessed 3 December 2021.