Food and Drug Administration Drug Approval Processes: Speedier Access to New Treatments
Caitlin N. Swann, PharmD
PGY2 Hematology/Oncology Pharmacy Resident
Cleveland Clinic, Cleveland, OH
Recently, there have been changes in the U.S. Food and Drug Administration’s (FDA’s) drug approval process to make important therapies available to patients sooner than ever before.1 This article reviews the various pathways developed by the FDA to expedite the drug approval process. Specifically, priority review, accelerated approval, fast-track designation, and the breakthrough therapy designation will be discussed along with examples of drugs that have gone through these programs.
Priority Review Versus Standard Review
To be eligible for priority review, a drug must treat a serious condition and provide a significant improvement in safety or effectiveness compared to standard treatment.1 A major benefit of this designation is that the FDA will review and make a decision on the drug approval application within 6 months, compared with 10 months under the standard review process. Those drugs granted priority review will also receive additional attention and resources from the FDA during the evaluation phase.
Accelerated Approval
The advantage of undergoing accelerated approval is that a drug may be approved based on a surrogate endpoint that is likely to predict a clinical benefit without having to demonstrate the clinical benefit itself.1 With accelerated approval, drugs can be approved much faster than the time it would take to demonstrate the drug’s impact on morbidity or mortality. To qualify for accelerated review, the drug must be used for a serious or life-threatening condition for which acceptable treatments are lacking. In conjunction with accelerated approval, the FDA requires the sponsors to agree to conduct post approval studies to verify that a clinical benefit has been demonstrated. If the study results confirm a clinical benefit, the FDA will convert the accelerated approval to traditional approval. If these studies fail to demonstrate a clinical benefit, the FDA may withdraw their approval of the agent, as was the case with ponatinib.
Ponatinib (Iclusig®), which is a tyrosine kinase inhibitor indicated for the treatment of chronic myeloid leukemia (CML) and Philadelphia positive (Ph+) acute lymphocytic leukemia (ALL) resistant or intolerant to other tyrosine kinase inhibitors, was approved in December 2012.2 During the initial phase 2 approval trial of this agent, a complete cytogenetic response was demonstrated in 46% of patients, while a major cytogenetic response was shown in 56% of patients, and a major molecular response was observed in 34% of patients.3 In addition, a subset of patients with the T315I mutation demonstrated even greater benefits from the drug, with 70% achieving a major cytogenetic response.3 The presence of the T315I mutation confers resistance to all previously tested tyrosine kinase inhibitors; ponatinib was shown to be the exception. Approval of ponatinib would provide a much needed treatment option for patients with this particular genetic mutation.
The FDA granted accelerated approval of ponatinib with the understanding that additional studies would be performed to confirm its benefit on morbidity and mortality and to further evaluate its safety.4At the time of approval, the initial study results demonstrated a favor- able risk profile for ponatinib. Unfortunately, as the data matured, the frequency of serious and life-threatening blood clots and severe narrowing of blood vessels increased from 9% in the initial reports to at least 27% in the most recent results.5 The FDA suspended the marketing and sales of ponatinib on October 31, 2013. During this time, the FDA analyzed the potential benefit in the subset of patients in whom this agent may still possess a favorable risk/ benefit ratio, specifically those with the T315I mutation. Ponatinib was not commercially available but could be obtained through a patient-specific investigational new drug (IND) or expanded access registry program. In December 2013 the FDA announced the reauthorization of marketing and sales of the drug with several new safety measures in place. A revised Risk Evaluation and Mitigation Strategy (REMS) highlights the cardiovascular risks associated with ponatinib as well as the new indications for its use. The indications are now limited to specific groups of patients: adults with T315I-positive CML or Ph+ ALL, and adults with chronic, accelerated, or blast-phase chronic myeloid leukemia or Ph+ ALL when no other tyrosine kinase inhibitor therapy is indicated. The commercial distribution of the drug resumed in mid-January 2014. Ponatinib is an example of how the accelerated approval process is meant to provide patients access to potentially life-saving treatments as soon as possible, while at the same time protecting the public from situations in which the actual risks outweigh the perceived benefits of treatment.
Fast Track Designation
The fast track designation also promotes expedited approval for drugs.6 In this program, sponsors of the drug will have frequent interactions with the FDA to discuss the drug’s development plan and field any questions about the process. If the FDA grants a drug fast track designation status, the sponsor may file certain portions of the marketing application before submitting the complete application in a process known as rolling review. Fast track designation may be requested at any stage during drug development. To qualify for the fast track designation, the drug must be intended for the treatment of a serious or life-threatening disease or condition and it must demonstrate the potential to address unmet medical needs for the intended disease or condition. An unmet medical need exists when available therapy does not address the treatment or diagnosis of a condition. The defining criteria or type of information needed to prove the drug addresses an unmet medical need depend on how far along in the process this expedited status is requested. For example, in nonclinical models, the rationale behind the mechanism of action or other pharmacologic data may be considered sufficient if the drug is early in the development phase. In contrast, if the drug is in the later stages of development and there are clinical data available, then this clinical data should be used to justify the potential to address an unmet need.
Breakthrough Therapy Designation
Breakthrough therapy designation is the newest FDA drug approval category, which was signed into law in 2012.6 This designation provides all of the same features that the fast track designation does but also allows for added guidance from the FDA. For drugs granted this designation, the FDA forms a multidisciplinary team that meets with the sponsor of the drug to provide advice in designing trials that will gather the necessary data efficiently to expedite the commercial approval of the drugs. In addition, the FDA will assign a senior manager to the approval application, along with a crossdisciplinary project lead from their review team to act as a liaison with the sponsor throughout the development process.
For a drug to receive the breakthrough therapy designation, the drug must meet certain criteria.6 It must be used to treat a serious condition or disease associated with morbidity and mortality, and it must have the potential to have a substantial impact on day-to-day functioning. In contrast to the fast track designation, this designation does not require an absence of available treatments for the disease or condition. Ideally, this designation can be used for drugs that demonstrate substantial improvements over existing therapies when measured using one or more clinically significant endpoints. These endpoints refer to those that measure effects on morbidity or mortality or on the presence and severity of symptoms caused by the disease. This is in contrast to the various forms of nonclinical information (e.g., theoretical or mechanistic rationale, early nonclinical data) required for fast track designation.
This new program is beginning to demonstrate an impact on drug development. In January 2013, the first two drugs to receive breakthrough designations were announced.1 Both designations were awarded to Vertex Pharmaceuticals, which was seeking to expand the use of their cystic fibrosis drug ivacaftor (Kalydeco®). As of November 8, 2013, 92 requests for breakthrough designations had been made, of which 30 were granted, 47 were denied, and 15 were still under review. The first drug with the breakthrough designation to be granted FDA approval was obinutuzumab (Gazyva®). It was approved on November 1, 2013, for use in combination with chlorambucil in patients with treatment-naïve chronic lymphocytic leukemia (CLL).7 The initial study that led to FDA approval of this drug was a phase 3, open-label study of 356 patients with previously untreated CLL.7,8 The study reported improvements in the primary endpoint of progression-free survival, which was 23 months with obinutuzumab plus chlorambucil versus 11.1 months with chlorambucil alone (p < .0001). The overall response rate was increased in the combination arm when compared with chlorambucil alone (76% versus 32.1%). Last, the median duration of response was 15.2 months versus 3.5 months, favoring the combination arm. The most common grade 3 or 4 adverse events for patients who received obinutuzumab in combination with chlorambucil compared with chlorambucil alone were infusion-related reactions during the first infusion (21% versus 0%), thrombocytopenia (11% versus 3%), and neutropenia (34% versus 16%), although the higher incidence of neutropenia did not correspond with an increased rate of infections in the obinutuzumab plus chlorambucil arm.
Following obinutuzumab’s approval, another drug received FDA approval under the accelerated approval program after being granted breakthrough therapy designation. Ibrutinib (Imbruvica®) received FDA approval on November 13, 2013, for patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.9 The drug made it through FDA review in just 4 months. Accelerated approval was granted based on the results of a phase 2 study that enrolled 111 previously treated patients with MCL.9,10 The primary endpoint was the overall response rate, with 66% of patients demonstrating this. Furthermore, 17% of patients achieved a complete response and 49% of patients achieved a partial response. The median duration of response was 17.5 months. Bleeding events, including bruising of any grade, occurred in 48% of patients, and 5% of these were grade 3 or higher. Treatment- emergent grades 3 or 4 cytopenias occurred in 41% of patients and 25% had grades 3 or 4 infection. As a condition of accelerated approval, the FDA required that the sponsor submit 24-month follow-up data for all patients in this single-arm trial. The sponsor must also submit the results of a randomized controlled trial comparing ibrutinib in combination with bendamustine plus rituximab to bendamustine plus rituximab alone in patients with newly diagnosed MCL.
Summary
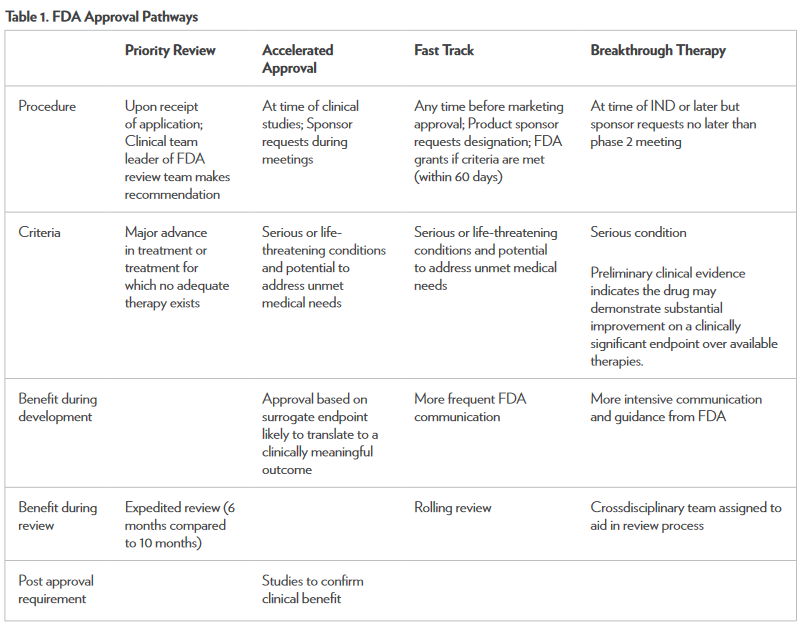
There are several FDA programs available to expedite the process of drug development and review to make potentially life-saving treatment options available to patients sooner.1,6 Along with the benefits of these programs, there are limitations. It is possible for a drug to make it to market before it has demonstrated safety and effectiveness. According to the FDA, there are stringent measures in place that require aggressive review from multidisciplinary teams representing both the FDA and the drug sponsor.6 The key component necessary for all of these programs is effective and timely communication between the sponsor and the FDA. The benefits of these programs have been demonstrated with the expedited approvals of drugs such as obinutuzumab and ponatinib, which were both made available to patients in a relatively short time. Table 1 summarizes the unique aspects of each approval pathway.3 Any healthcare professional can and should report adverse effects caused by any medication—new or old—through the MedWatch section on the FDA’s website.11 The new FDA approval pathways have the potential to greatly impact the treatment of cancer. Drugs are being approved faster than ever, meeting the needs of many cancer patients.
References
1. Fast track, breakthrough therapy, accelerated approval and priority review. Expediting availability of new drugs for patients with serious conditions. U.S. Food and Drug Administration website. www.fda.gov/forconsumers/ byaudience/forpatientadvocates/speedingaccesstoimportantnewtherapies/ucm128291.htm. Updated June 26, 2013. Accessed December 13, 2013.
2. Iclusig™ (ponatinib) [package insert]. Cambridge, MA: ARIAD, Inc; December 2012.
3. Cortes E, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome–positive leukemias. N Engl J Med. 2013;369:1784-96.
4. Approved drugs: Ponatinib. U.S. Food and Drug Administration website. www.fda.gov/ Drugs/InformationOnDrugs/ApprovedDrugs/ucm332368.htm. Updated December 17, 2012. Accessed December 13, 2013.
5. FDA drug safety communication. U.S. Food and Drug Administration website. www.fda.gov/Drugs/ DrugSafety/ucm379554.htm. Updated January 7th, 2014. Accessed January 10, 2014.
6. Guidance for industry expedited programs for serious conditions–drugs and biologics. U.S. Food and Drug Administration website. www.fda.gov/downloads/ Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM358301.pdf. Updated June 2013. Accessed December 13, 2013.
7. Gazyva® (obinutuzumab) [package insert]. South San Francisco, CA: Genentech, Inc; November 2013.
8. Goede V, Fischer K, Humphrey K, et al. Obinutuzumab (GA101) plus chlorambucil (Clb) or rituximab (R) plus Clb versus Clb alone in patients with chronic lymphocytic leukemia (CLL) and preexisting medical conditions (comorbidities): Final stage 2 results of the CLL11 (BO21004) phase III trial [abstract]. Blood. 2013;122:6
9. Approved drugs: Ibrutinib. U.S. Food and Drug Administration website. Available at: ww.fda.gov/ Drugs/InformationOnDrugs/ApprovedDrugs/ucm374857.htm. Updated November 13, 2013. Accessed December 13, 2013.
10. Wang ML, Rule SR, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507-16.
11. Reporting serious problems to FDA. U.S. Food and Drug Administration website. www.fda.gov/ Safety/MedWatch/HowToReport/default.htm. Updated August 23, 2013. Accessed December 30, 2013.